南昌航空大学陈德志课题组ESE:氧空位引导苯酚表面聚合,推动高级氧化净水低药耗精准转化
- 2026-06-17 11:07:05


论文的视频解读由NotebookLM AI智能生成,内容仅供参考,具体研究请查阅原文。
导语
高级氧化过程是去除水中难降解有机污染物的重要技术路径,已广泛用于处理药物残留、个人护理品、农药及其他持久性有机污染物。传统体系通常依赖硫酸根自由基(SO4⦁−)和羟基自由基(⦁OH)等高活性物种,氧化能力强,但也常伴随较高氧化剂消耗、潜在毒性中间产物生成以及运行碳排放增加等问题。
如何让污染物“消失”得更高效,同时减少无效氧化和副产物风险,是高级氧化技术走向绿色低碳应用必须解决的关键问题。
近日,Environmental Science and Ecotechnology发表题为“Oxygen vacancy–rich spinel oxide drives phenol polymerization via direct oxidative transfer”的原创研究论文。来自南昌航空大学、南开大学、北京航空航天大学等机构的研究人员以苯酚为模型污染物,构建了氧空位富集的锰铁尖晶石氧化物/碳布催化体系,发现氧空位能够调控过一硫酸盐(peroxomonosulfate, PMS)活化路径,推动苯酚通过催化剂表面限域的直接氧化转移过程(direct oxidation transfer process, DOTP)发生聚合,而非主要依赖自由基主导的水相无选择性氧化。
研究表明,氧空位不仅提高了催化活性,更改变了污染物的转化命运。通过提高氧空位密度,催化剂可抑制自由基生成,促进污染物与氧化剂在表面发生两电子直接氧化还原转移,使苯酚转化为富集在催化剂表面的聚合产物。在连续流反应器中,优化催化剂在240 h运行中保持97.5%的苯酚去除率和73.2%的总有机碳去除率,同时金属离子浸出低于0.2 mg L−1。
这一研究的核心意义在于,它将“缺陷工程”从提高催化剂活性,进一步推进到调控污染物反应路径。对于水处理高级氧化技术而言,未来评价催化剂性能不能只看污染物去除速度,还应关注污染物最终转化为何种形态、是否形成难控副产物,以及氧化剂是否真正用于有效转化。
高级氧化过程需要从“强反应”走向“可控转化”
01
高级氧化过程之所以受到广泛关注,是因为其能够通过高活性氧化物种快速降解常规生物处理难以去除的有机污染物。但自由基氧化也存在天然局限:自由基反应活性高、选择性弱,容易同时攻击目标污染物、天然有机物和水体背景组分;污染物在逐步氧化过程中还可能形成毒性更强或更难去除的中间产物。
近年来,非自由基路径受到越来越多关注。其中,直接氧化转移过程(DOTP)提供了一种不同思路:污染物和氧化剂不再主要依赖水相自由基扩散反应,而是在非均相催化剂表面同时被活化,通过直接电子转移形成表面中间体,并进一步发生聚合或偶联反应。如此,有机污染物并非完全沿着碎片化降解路径进入水相,而是可被转化并富集在催化剂表面,从而实现低氧化剂消耗条件下的有效去除。
这一路径的难点在于“可控”。同样是PMS活化,不同催化剂可能导向自由基氧化、单线态氧路径、高价金属氧物种路径,也可能导向DOTP。如何设计催化剂,使其优先推动污染物发生表面限域的两电子转移和聚合反应,是该领域的重要科学问题。
本研究选择锰铁尖晶石氧化物(MnFe2O4)作为催化剂主体,并通过在氮气气氛下热处理引入氧空位。尖晶石氧化物具有较高结构稳定性和可调控的金属位点,氧空位则可改变局部电荷分布、暴露活性位点并调节电子转移过程。研究团队由此构建了不同氧空位浓度的MnFe2O4/碳布自支撑催化剂,系统考察氧空位如何决定PMS活化方式和苯酚转化路径。
氧空位密度升高,反应路径随之改变
02
研究团队首先通过不同温度退火制备了一系列MnFe2O4/碳布催化剂,分别记为MFOCC400、MFOCC500、MFOCC600、MFOCC700和MFOCC800。随着退火温度升高,催化剂晶格间距逐渐减小,X射线衍射峰向高角度偏移,电子顺磁共振信号增强,表明表面氧空位浓度持续升高。原位高温拉曼光谱也进一步证明,热处理可在Fe和Mn位点附近诱导结构缺陷形成。
在苯酚/PMS体系中,所有MFOCC催化剂均可在55 min内实现苯酚完全去除。更值得关注的是,总有机碳去除率随氧空位浓度升高而提高,从MFOCC400体系的71.2%提升至更高氧空位样品中的约85%。这说明氧空位不仅加快污染物消失,也提高了有机碳从水相中去除的程度。
但这并不意味着污染物完全被矿化为CO2和H2O。COD分布分析显示,在MFOCC/PMS/苯酚体系中,反应后约70%~85%的初始COD转移到催化剂表面,而非继续残留在水相中。无PMS对照实验中,催化剂对苯酚的吸附贡献有限,说明这种表面有机碳富集不是简单物理吸附,而是反应生成物在催化剂表面的积累。XPS分析进一步显示,反应后催化剂表面C–O和C–O–C键含量增加,为表面聚合产物形成提供了直接证据。
由此,氧空位的作用已经超出传统意义上的“提高活性”。它改变了污染物的反应路线,使苯酚在催化剂表面被活化、转移、聚合并固定。对于高级氧化过程而言,这意味着污染控制可以从“尽可能强烈地氧化”转向“有选择地引导转化”。
苯酚在催化剂表面发生限域聚合
03
为确认苯酚的转化形态,研究团队对反应后催化剂表面产物进行了红外光谱、热重分析、MALDI-TOF质谱、凝胶渗透色谱、热重-红外-气质联用和核磁共振等多重表征。
傅里叶变换红外光谱显示,反应后催化剂表面出现约1200 cm−1的C–O–C特征峰,指向聚苯醚类结构形成。研究进一步区分了两类表面反应路径:一类是C–O聚合反应路径,生成聚合产物;另一类是C–C偶联反应路径,生成小分子偶联产物。随着氧空位浓度升高,乙醇洗脱得到的小分子偶联产物颜色逐渐变浅,而甲苯洗脱和残留COD分析显示聚合产物比例增加,说明氧空位会抑制偶联路径、促进聚合路径。
为简化产物分析,研究团队以2,6-二甲基苯酚替代苯酚开展实验。由于2,6-二甲基苯酚只有一个活性氢位点,更适合追踪线性聚合。质谱和凝胶渗透色谱结果显示,反应后催化剂表面形成线性聚苯醚产物,中心分子量约为3513 Da,多分散系数为2.4。热重-红外-气质联用和13C核磁共振也进一步确认了聚苯醚结构。
这一结果具有重要启示:污染物去除并非只有“完全矿化”一种理想终点。在特定催化界面上,将污染物转化为可从水相中分离、可在催化剂表面富集的聚合物,也可能成为一种低药耗、低副产物风险的污染控制策略。
图1解读
图1集中展示了催化剂表面产物的关键证据。红外光谱中C–O–C特征峰表明聚苯醚结构形成;乙醇洗脱液颜色随退火温度升高而变浅,说明小分子偶联产物减少;COD分布进一步显示,聚合产物贡献随氧空位浓度增加而提高。MALDI-TOF质谱则确认,2,6-二甲基苯酚可在MFOCC800表面形成线性聚苯醚产物。
图1说明苯酚并非简单在水相中被自由基逐步破碎,而是在催化剂表面发生有组织的聚合和偶联转化。氧空位浓度越高,反应越偏向聚合路径。
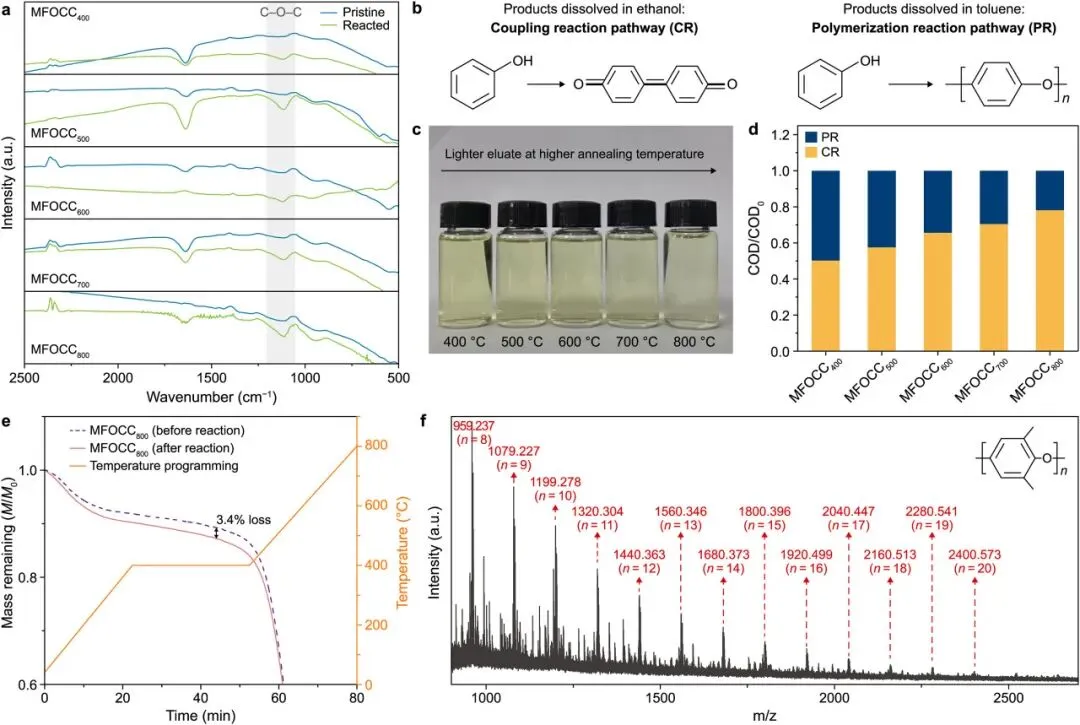
图1 反应后MFOCC催化剂上偶联和聚合的光谱及产物证据。a,不同MFOCC-VO催化剂在原始状态和反应后的傅里叶变换红外光谱。灰色阴影区域表示C–O–C键的信号峰。b,偶联反应(CR)和聚合反应路径(PR)的示意图。c,反应后洗脱小分子产物之后的乙醇洗脱液光学照片;如箭头所示,洗脱液随退火温度升高逐渐变浅。d,反应后MFOCC-VO上不同洗脱产物的化学需氧量(COD)分布。e,原始MFOCC800和反应后MFOCC800在空气中记录的热重曲线;橙色线表示升温程序。f,用甲苯洗脱产物的基质辅助激光解吸/电离飞行时间质谱图。插图显示聚(2,6-二甲基-1,4-苯醚)(PPO)聚合单元的示意图;一个单元的质荷比可粗略估算为120.1;标注数值为相应峰的质荷比,n表示聚合度。实验条件:[PhOH]COD = 0.15 g L−1,[catalysts]COD = 1.5 g L−1,[PMS]COD = 0.75 g L−1,30 ℃。PMS,过一硫酸盐;VO,氧空位。MFOCC400–MFOCC800分别表示在400~800 ℃退火的样品。
两电子直接转移构成非自由基路径的关键证据
04
为阐明DOTP的反应机制,研究团队考察了PMS与苯酚的投加比例、反应动力学和界面电子转移过程。结果显示,PMS与苯酚的最佳化学计量比约为5:1;在该比例下,反应呈现二级动力学特征,明显不同于传统活性氧物种主导高级氧化过程中常见的准一级动力学行为。
更关键的是,研究设计了双室和单室两类反应系统。在双室系统中,PMS和苯酚通过外电路实现电子联系,但污染物与催化剂表面不能充分接触。此时,虽然电流响应随氧空位浓度增加而增强,苯酚去除效率却下降。相反,在单室系统中,PMS、苯酚和催化剂处于同一反应界面,MFOCC800可在60 min内实现近100%的苯酚去除。该结果说明,DOTP并不是单纯远程电子转移即可完成的过程,污染物与氧化剂必须在催化剂表面发生直接接触和协同活化。
研究还以碘化钾作为典型两电子转移试剂进行对照。碘化钾可将PMS还原为SO42−,同时不产生SO4⦁−或•OH。实验观察到相似的去除趋势,支持苯酚去除遵循两电子转移路径。EPR自旋捕获结果未检测到DMPO-OH或DMPO-SO4加合物,自由基淬灭和探针测试也进一步确认该过程具有非自由基特征。
此外,乙腈-水混合溶剂显著抑制反应,表明苯氧自由基中间体参与了后续转化;以D2O替代H2O时,体系出现明显二级动力学同位素效应,说明反应过程中苯酚邻位碳原子的杂化状态发生变化。综合这些结果,研究提出:苯酚与活化PMS在氧空位位点发生两电子转移,生成表面苯氧自由基中间体,随后在催化剂表面进一步发生C–O聚合或C–C偶联反应。
图2解读
图2从反应计量、反应动力学和界面电子转移三个角度说明DOTP机制。PMS/苯酚投加比为5时,体系呈现二级动力学;双室和单室反应对比显示,污染物与催化剂表面的直接接触对于高效去除至关重要。碘化钾对照实验和EPR结果进一步支持两电子转移和非自由基路径。
图2的价值在于将“非自由基”判断从现象层面推进到机制层面:MFOCC800不是简单产生更多自由基,而是提供了一个表面反应平台,使PMS和苯酚在界面上发生定向氧化还原转移。
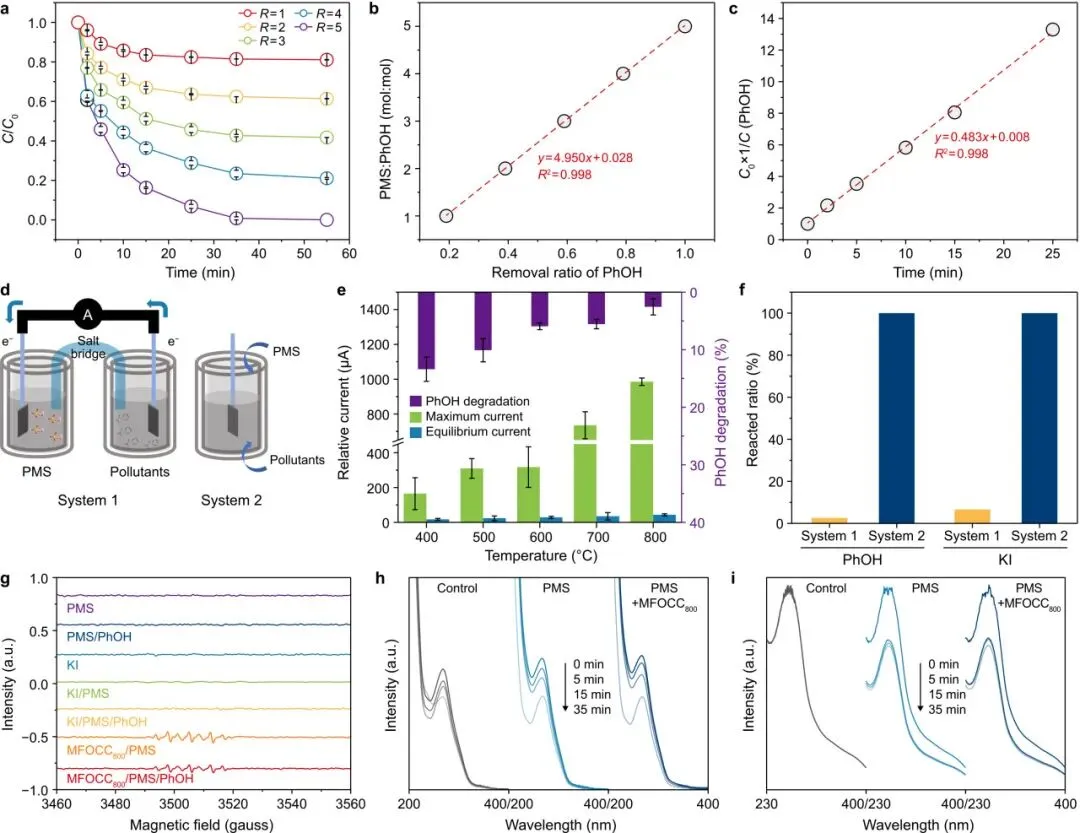
图2 直接氧化转移对PMS/PhOH比例的依赖性以及界面电子转移路径的证据。a,不同PMS与苯酚(PhOH)投加比例下,MFOCC800/PMS体系中的苯酚去除曲线。R = CPMS/CPhOH,其中CPMS和CPhOH分别为PMS和PhOH浓度。b,R与PhOH去除效率之间的相关关系。c,R = 5时反应的二级动力学拟合。d,双室(系统1)和单室(系统2)反应系统示意图。e,不同双室反应设置中的电流响应和PhOH去除效率变化。f,MFOCC800基体系中PhOH和KI的转化。g,不同体系中以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)作为自旋捕获剂的EPR光谱。h–i,不同体系中1,3-二苯基异苯并呋喃(DPBF;h)和氯化硝基蓝四唑(NBT;i)的时间依赖紫外-可见光谱。实验条件:[PhOH] = 0.01 g L−1,[catalysts] = 0.1 g L−1,[NBT] = 10 mmol L−1,[DPBF] = 0.03 mmol L−1,[PMS] = 0.05 g L−1,30 ℃。PMS,过一硫酸盐;MFOCC800,800 ℃退火样品。
电子结构重塑让PMS与苯酚更易协同活化
05
实验揭示了反应路径,理论计算进一步解释了氧空位驱动DOTP的原因。
密度泛函理论计算显示,在无氧空位的MFOCC中,电荷主要局域在氧位点,分布相对均匀且受限;引入氧空位后,电子向邻近氧位点重新分布,形成明显的电子离域状态。这种电子结构变化有利于PMS和苯酚在催化剂表面富集并被同时活化。
与原始MFOCC相比,MFOCC-VO的价带边缘从2.82 eV上移至2.43 eV,带隙减小,电子转移阻力降低;d带中心也从−1.10 eV上移至−0.95 eV,占据态电子数由0.40增至0.43。这些变化增强了催化剂对PMS的化学吸附能力,PMS吸附能由−2.44 eV增强至−4.09 eV。
PMS吸附后的键长变化同样支持该机制。在MFOCC上,PMS中的O–O键仅由1.479 Å拉长至1.514 Å;而在MFOCC-VO上,O–O键显著拉长至2.778 Å,显示氧空位强烈促进PMS的O–O键断裂,并生成SO42−和OH−,而不是诱导自由基路径。
分子动力学模拟进一步显示,在无氧空位的MFOCC体系中,PMS和苯酚分子更容易向外扩散;而在MFOCC-VO体系中,PMS和苯酚逐渐聚集在催化剂表面。这说明氧空位不仅改变电子结构,也提供了表面限域环境,使氧化剂和污染物在界面上更容易发生定向两电子转移。
图3解读
图3展示了氧空位影响催化反应的理论计算证据。引入氧空位后,催化剂表面出现明显电子离域,价带边缘上移,d带中心正移,PMS吸附增强,分子在催化剂表面扩散受限。自由能分析还表明,MFOCC-VO更倾向于稳定聚合路径,同时抑制传统自由基高级氧化路径。
图3是理解“氧空位为什么能改变反应路径”的关键。氧空位并非只提供更多活性位点,而是重塑催化剂表面的电子环境,使其同时优化PMS还原活化和苯酚氧化活化,最终将反应导向表面限域DOTP。
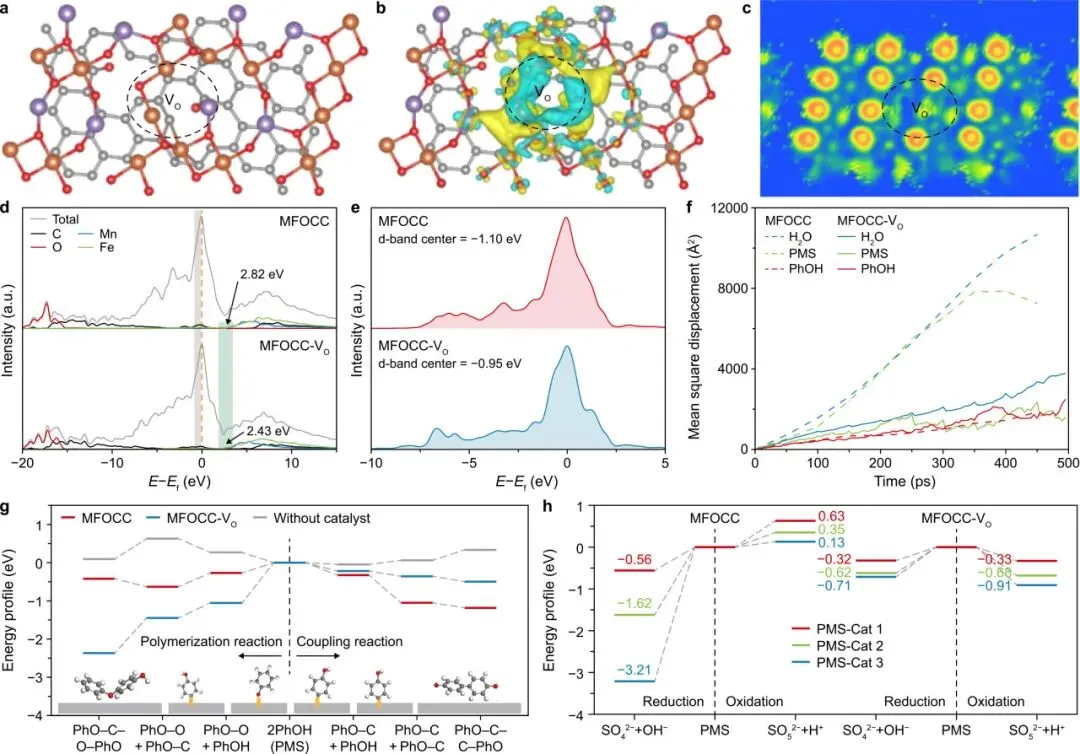
图3 MFOCC中氧空位作用的理论和光谱学认识。a–c,MFOCC-VO的计算模型(a)、差分电荷密度图(b)和电子局域函数图(c)。黄色表示电子积累,蓝色表示电子耗尽。d–e,MFOCC和MFOCC-VO的态密度(DOS;d)以及d轨道分波态密度(PDOS;e)。在d图中,灰色阴影表示占据态,绿色阴影表示价带边缘,橙色虚线表示费米能级。f,MFOCC和MFOCC-VO的均方位移。g,不同体系中直接氧化转移过程(DOTP)的自由能垒。灰色方块表示催化剂表面,黄色标记表示吸附态/中间体。h,高级氧化过程中催化剂表面过一硫酸盐(PMS)氧化和还原活化的自由能垒。VO,氧空位。
从MnFe2O4到Mn3O4和α-FeOOH,缺陷调控规律得到跨体系验证
06
为检验氧空位调控DOTP的适用范围,研究团队将这一思路扩展到Mn3O4和α-FeOOH两类典型氧化物催化剂中。
在Mn3O4/PMS体系中,苯酚去除效率随氧空位浓度变化而变化。氧空位含量最高的Mn3O4-VO1体系实现79.8%的TOC去除,高于Mn3O4-VO2的77.6%和无氧空位Mn3O4-VO0的43.2%。COD分析显示,表面聚合产物贡献随氧空位含量升高而增加。
α-FeOOH体系中也观察到类似规律。随着表面氧空位浓度升高,苯酚去除效率由51.4%提升至99.2%,反应速率常数由0.017 min−1增至0.114 min−1;55 min后的TOC去除率由42.9%提升至82.3%。同样,反应后表面COD和聚合产物比例均随氧空位浓度增加而提高。
这些结果说明,氧空位促进DOTP和聚合路径并非MnFe2O4体系中的偶然现象,而可能是氧空位富集氧化物催化剂中具有一定普适性的结构—活性关系。对于催化剂设计而言,这为通过缺陷工程定向调控有机污染物转化路径提供了可推广原则。
真实水体和连续流运行检验工程适用性
07
面向实际水处理应用,催化体系必须经受水体基质、长期运行和多污染物条件的检验。研究团队选取优化后的MFOCC800催化剂,进一步评估其在不同水体和连续流反应器中的表现。
结果显示,MFOCC800/PMS体系对环境基质具有较强耐受性。NaCl、NaNO3、NaHCO3和腐殖酸对苯酚去除影响较小;在不同水体中,该体系仍可保持高效去除,去离子水中苯酚去除率接近100%,自来水中为99.5%,河水中为98.7%,二级出水中为97.3%。二级出水中效率略低,主要归因于天然有机物与苯酚对活性位点的竞争吸附。
催化剂循环性能同样良好。经去离子水、乙醇和甲苯依次清洗去除表面有机物后,MFOCC800在连续8次循环中仍可在55 min内保持95.1%的苯酚去除效率。
该体系对其他有机污染物也表现出较高去除能力,包括苯胺(98.5%)、磺胺甲噁唑(99.2%)、四环素(97.3%)、双酚A(98.7%)和罗丹明B(100%)。不过,不同污染物通过DOTP转化的贡献并不相同:双酚A最高,为80.3%;苯胺为63.9%;磺胺甲噁唑为49.9%;罗丹明B为41.1%;四环素为29.5%。这说明DOTP对污染物结构具有选择性,尤其有利于含有较多邻位或对位活性氢的芳香族化合物发生表面聚合或偶联。
连续流实验进一步展示了工程应用潜力。在240 h运行中,MFOCC800保持稳定性能,实现97.5%苯酚去除、73.2%TOC去除和最终69.7%COD去除;出水中Mn和Fe离子最高浓度分别为0.08 mg L−1和 0.16 mg L−1,表明催化剂结构稳定、金属浸出较低。
图4解读
图4展示了MFOCC800/PMS体系在实际水处理条件下的表现。无机盐和腐殖酸对苯酚去除影响较小;自来水、河水和二级出水中仍保持高去除率;8次循环后活性保持在95.1%;对多种有机污染物均表现出较高去除能力。连续流运行240 h后,体系仍保持高效苯酚和TOC去除,并且金属浸出较低。
图4将机理研究与应用场景连接起来。对于催化水处理技术而言,稳定性、抗干扰能力和连续运行表现,往往比单次批量实验更能说明其工程潜力。
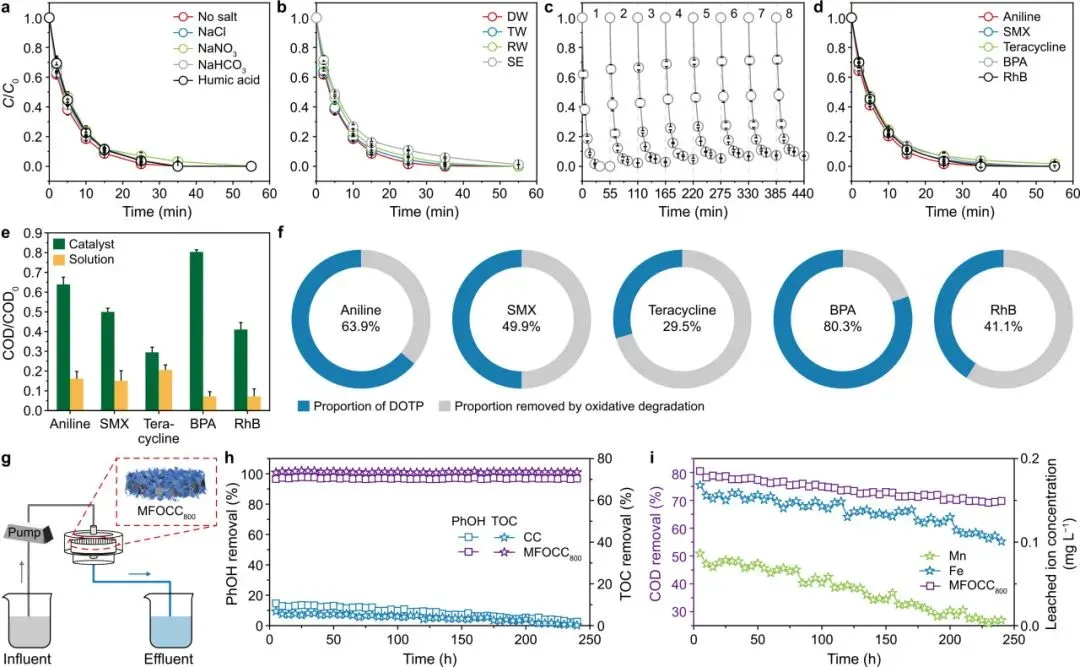
图4 MFOCC800/PMS体系的基质效应、可重复使用性、污染物适用范围和连续流性能。a,在无机盐和腐殖酸存在下,MFOCC800/PMS体系中的PhOH去除曲线。b,不同水体基质条件下的PhOH去除。c,MFOCC800/PMS体系1~8次循环的可重复使用性。d,MFOCC800/PMS体系对多种污染物的去除表现。e,不同污染物处理后反应溶液和催化剂表面的残余化学需氧量(COD)。f,不同污染物在去除过程中通过直接氧化转移过程(DOTP)积累在催化剂表面的比例。g,连续流检测过程示意图。h–i,连续流反应器中的PhOH和总有机碳(TOC)去除(h),以及相应的化学需氧量(COD)去除和金属离子浸出(i)。实验条件:[Pollutants] = 0.01 g L−1,[catalysts] = 0.1 g L−1,[PMS] = 0.05 g L−1,流速 = 2 mL min−1,30 ℃。PMS,过一硫酸盐;MFOCC800,800 ℃退火样品;DW,去离子水;TW,自来水;RW,河水;SE,二级出水;SMX,磺胺甲噁唑;BPA,双酚A;RhB,罗丹明B。
从“去除效率”转向“转化路径”设计
08
这项研究为高级氧化水处理提供了一个重要转向:污染物治理不应只追求更快的浓度下降,还应关注污染物在反应过程中走向何处。传统自由基氧化常以强氧化能力为优势,但其反应路径难以精确控制;DOTP则强调在催化剂界面上同时活化氧化剂和污染物,通过直接电子转移推动表面聚合或偶联,使污染物以更可控方式从水相中移除。
氧空位在这一过程中发挥了核心作用。它通过电子离域、d带中心调节和表面吸附增强,使PMS和苯酚在催化剂表面形成更有利的反应构型;同时,氧空位富集体系抑制自由基路径,降低了传统高级氧化过程中无选择性反应和副产物生成的风险。这意味着缺陷工程可以成为调控污染物化学命运的工具,而不仅是提高反应速率的手段。
从应用角度看,MFOCC800在真实水体、循环使用和连续流运行中保持稳定性能,表明该策略具有进一步工程验证价值。更重要的是,该氧空位依赖的结构—活性关系在Mn3O4和α-FeOOH等体系中得到验证,说明这一设计原则有望扩展到更多氧化物催化剂。
未来仍需进一步评估聚合产物的长期稳定性、催化剂再生方式、复杂废水中多污染物竞争行为,以及连续运行过程中的材料寿命和全流程环境影响。只有将反应路径控制、材料稳定性和工程经济性纳入统一框架,DOTP才可能从机理研究走向规模化水处理应用。
结语
水中有机污染物治理正在从“强氧化去除”走向“可控转化去除”。这项研究表明,氧空位富集的尖晶石氧化物能够在PMS体系中重塑苯酚氧化路径,使污染物通过表面限域直接氧化转移发生聚合,并在催化剂表面富集。其本质不是让自由基反应更猛烈,而是让界面电子转移更有方向、让污染物转化更可控。
通过调控氧空位密度,研究小组建立了缺陷结构与聚合路径贡献之间的关联,并在多种氧化物体系中验证了这一规律。优化催化剂在连续流中实现240 h稳定运行,保持高苯酚去除率和TOC去除率,显示出面向实际水处理的潜在价值。
面向低碳、低药耗和高选择性的水污染控制需求,这项研究提供了一条新的催化设计思路:通过原子尺度缺陷工程,不仅提高污染物去除效率,更精准决定污染物在处理过程中的化学去向。对于未来高级氧化技术的发展而言,这种从“反应强度”转向“路径选择”的理念,可能成为构建绿色净水体系的重要方向。
作者信息

第一作者:叶新春,南昌航空大学环境与化学工程学院2025届硕士研究生,目前博士就读于南开大学环境科学与工程学院,研究方向为高级氧化水处理技术,已在Environmental Science and Ecotechnology、Journal of Hazardous Materials等环境重要期刊参与发表多篇SCI论文。

通讯作者:陈德志,博士、南昌航空大学环境与化学工程学院教授,博士生导师,入选江西省主要学科学术和技术带头人培养计划及江西省科技创新杰出青年人才培养对象。长期致力于微纳材料的可控制备、界面调控及其在水体修复等领域的应用研究。发表SCI收录论文60余篇,H指数39;获授权中国发明专利10余项,相关成果曾获中国发明创业奖-创新奖一等奖。
课题组介绍:本课题组依托江西省持久性污染物控制与资源循环利用重点实验室,隶属于由罗旭彪教授与邹建平教授共同领衔的研究团队。团队长期聚焦退役动力电池资源化、废水治理与资源回收、光电催化能源存储等前沿方向,成果荣获国家科技奖及多项省部级一等奖。
课题组科研氛围浓厚,实验平台先进,科研经费充足。热忱欢迎优秀学子报考本课题组硕士、博士研究生,共同投身于环境与能源领域的关键技术突破。
引用信息
Ye, X., Wang, Y., Zhou, T., Hu, Y., Zou, L., Zou, Q., ... & Luo, X. (2026). Oxygen Vacancy–Rich Spinel Oxide Drives Phenol Polymerization via Direct Oxidative Transfer. Environmental Science and Ecotechnology 32: 100710.
doi: 10.1016/j.ese.2026.100710

来源:ESE期刊。投稿、合作、转载、进群,请添加小编微信Environmentor2020!环境人Environmentor是环境领域最大的学术公号,拥有25W+活跃读者。由于微信修改了推送规则,请大家将环境人Environmentor加为星标,或每次看完后点击页面下端的“在看”,这样可以第一时间收到我们每日的推文!环境人Environmentor现有综合群、期刊投稿群、基金申请群、留学申请群、各研究领域群等共20余个,欢迎大家加小编微信Environmentor2020,我们会尽快拉您进入对应的群。

往期推荐



扫描二维码,快速入群~
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 赣羽超|南昌四支队伍挺进2026年“赣羽超”八强!
- 燃HIPHOP之夜·南昌站|全员阵容公布,热血唱响赣鄱之夜!
- 原木奶油风,实木烤漆混油,南昌润府整木定制安装中,客户说隔壁邻居上下楼都过去看了,味道都好重,唯独我们家没什么味道,全部是实木基
- 【和美教育·学生活动】南昌县洪亿学校开展2026年庆六一“和美”校园艺术节活动
- 南昌:“一刻钟宠物生活圈”渐成型
- 热烈欢迎南昌市红谷滩区政协副主席周思全一行莅临我会座谈交流
- 【转载】2026中国南昌国际龙舟赛,出行攻略请查收!
- 【展商推荐】南昌市新威实业有限公司
- 南昌被低估的三甲医院!这家综合实力不逊热门附院,江西人建议西湖患者不必一味外求
- 江右望族|南昌洲渼李氏:源自唐代名将,千年西平文脉永续
