从菌群失衡到炎症失控
急性胰腺炎不仅关乎胰腺自身的损伤,更与肠道屏障的崩溃密切相关。炎症风暴会破坏肠道完整性,导致细菌及其产物易位,进一步加剧全身炎症,形成恶性循环。此前研究已发现,患者肠道内双歧杆菌等有益菌显著减少,但其具体保护机制仍不清晰。研究团队从临床观察出发,将目光投向了双歧杆菌假长亚种——一种在人和哺乳动物肠道中常见的益生菌。
团队首先在小鼠模型中进行了验证。通过雨蛙肽注射或胰管结扎两种方法分别诱导轻度和重度胰腺炎模型,并在建模前给予小鼠口服该益生菌。结果显示,益生菌预处理能显著减轻胰腺组织的水肿、坏死和炎症细胞浸润,同时降低血清中淀粉酶和脂肪酶水平。更重要的是,它减少了胰腺组织中促炎的M1型巨噬细胞标记物表达,并提升了抗炎的M2型巨噬细胞标记物。
代谢物是关键,而非细菌本身
一个关键问题是:益生菌是如何远程发挥作用的?是细菌本身迁移到了胰腺吗?检测结果给出了否定答案。无论是在胰腺还是肝脏组织中,都未发现双歧杆菌假长亚种的直接踪迹。这提示,其保护作用可能源于其分泌的某些活性分子。
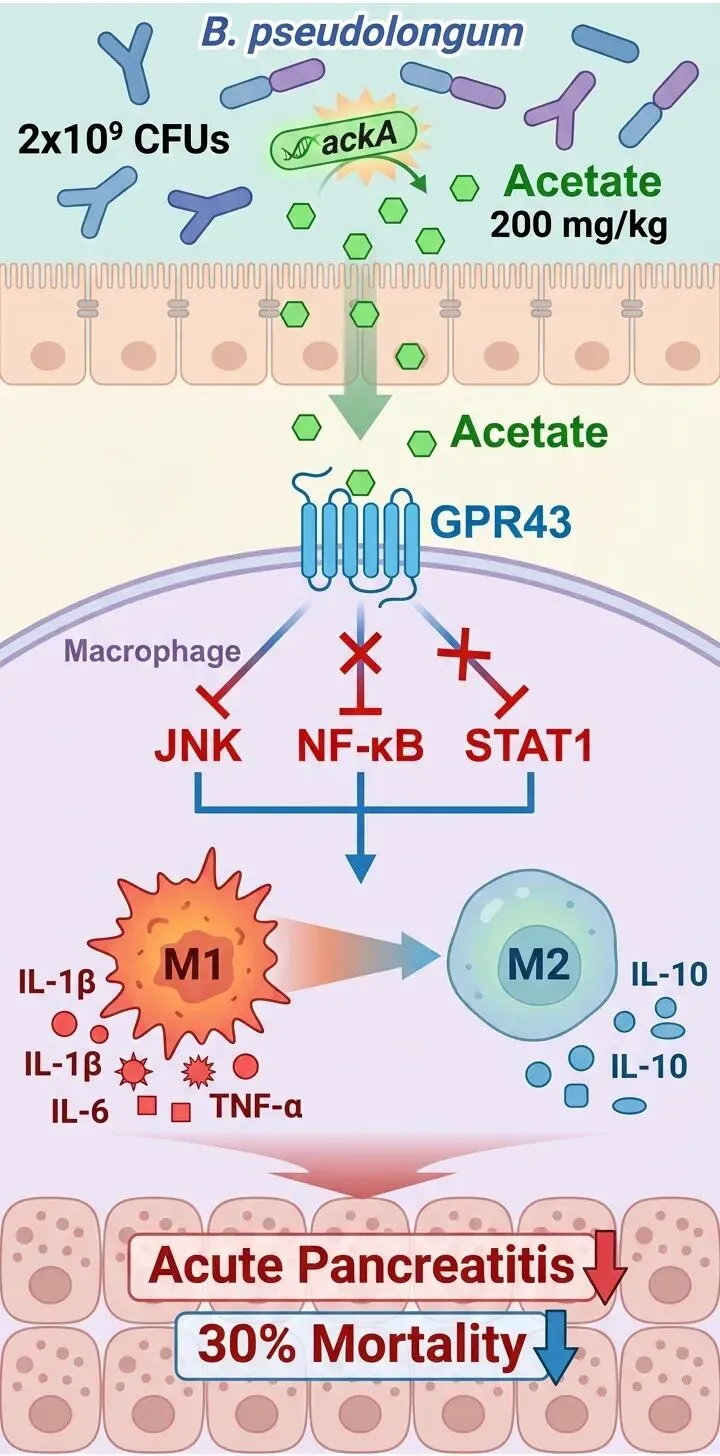
为了证实这一点,团队比较了活菌与热灭活死菌的效果。只有活菌能有效缓解胰腺炎,而死菌的保护作用几乎丧失。这强烈指向了细菌代谢产物的核心地位。随后的代谢组学分析将目标锁定在短链脂肪酸,特别是其中含量最丰富的乙酸上。无论是在细菌培养上清还是无菌小鼠的粪便中,乙酸水平都因该益生菌的存在而显著升高。
基因“开关”与受体的精妙对话
细菌如何产生乙酸?全基因组测序找到了关键基因ackA(乙酸激酶基因)。实验表明,该基因的活性在活菌中保持,而在热灭活菌中则明显降低。为了直接证明ackA的功能,研究人员进行了一项巧妙的“基因移植”实验:将双歧杆菌的ackA基因导入本身缺失该基因的工程化大肠杆菌中。结果,这株“改造后”的大肠杆菌产生了与双歧杆菌相当的乙酸水平,并在小鼠胰腺炎模型中重现了类似的保护效果。
乙酸又如何将信号传递给免疫系统?已知短链脂肪酸主要通过一类名为G蛋白偶联受体的蛋白发挥作用,其中GPR43是乙酸的主要受体。当研究使用GPR43特异性拮抗剂或通过病毒载体在胰腺特异性敲低该受体后,乙酸的保护作用便被完全阻断。胰腺损伤、炎症细胞浸润和细胞凋亡均恢复至严重水平。这证实了乙酸-GPR43这条信号轴的必要性。
驯服巨噬细胞,扭转炎症极化
那么,激活GPR43后,究竟改变了什么?对胰腺组织的单细胞测序数据分析显示,GPR43在髓系免疫细胞(包括巨噬细胞)中高表达。在胰腺炎早期,大量巨噬细胞浸润胰腺,并极化为促炎的M1表型,分泌白介素-1β、肿瘤坏死因子-α等因子,驱动炎症恶化。
研究发现,无论是给予益生菌还是直接补充乙酸,都能显著减少胰腺和脾脏中巨噬细胞的总数,并抑制其向M1型极化,同时促进其向具有修复功能的M2型转化。当使用药物清除体内的巨噬细胞后,乙酸的保护作用便消失了,这直接证明了巨噬细胞是乙酸发挥作用不可或缺的“执行者”。
在分子层面,乙酸通过GPR43,抑制了巨噬细胞内三条关键的促炎信号通路:JNK、NF-κB和STAT1的激活。这三条通路好比炎症反应的“加速踏板”,乙酸的介入有效地踩下了刹车。
从实验室到临床的呼应
研究的最后一部分回到了临床。团队分析了急性胰腺炎患者与健康人的粪便样本,发现患者粪便中双歧杆菌假长亚种的丰度和乙酸水平均显著下降,且在病情更重的患者中降低更为明显。这些指标与血液中的炎症标志物(如C反应蛋白) 呈负相关。这为实验室发现提供了有力的人类证据支持。
综上所述,这项研究描绘了一条清晰的通路:双歧杆菌假长亚种在肠道内定植并产生乙酸,乙酸通过血液循环或其他途径抵达胰腺,激活巨噬细胞上的GPR43受体,进而抑制细胞内促炎信号,阻止巨噬细胞向有害的M1型极化,最终缓解胰腺炎症和组织损伤。这不仅深化了我们对“肠-胰轴”互作的理解,也为开发以特定益生菌或其代谢产物为基础的急性胰腺炎预防与治疗新策略提供了坚实的科学依据。