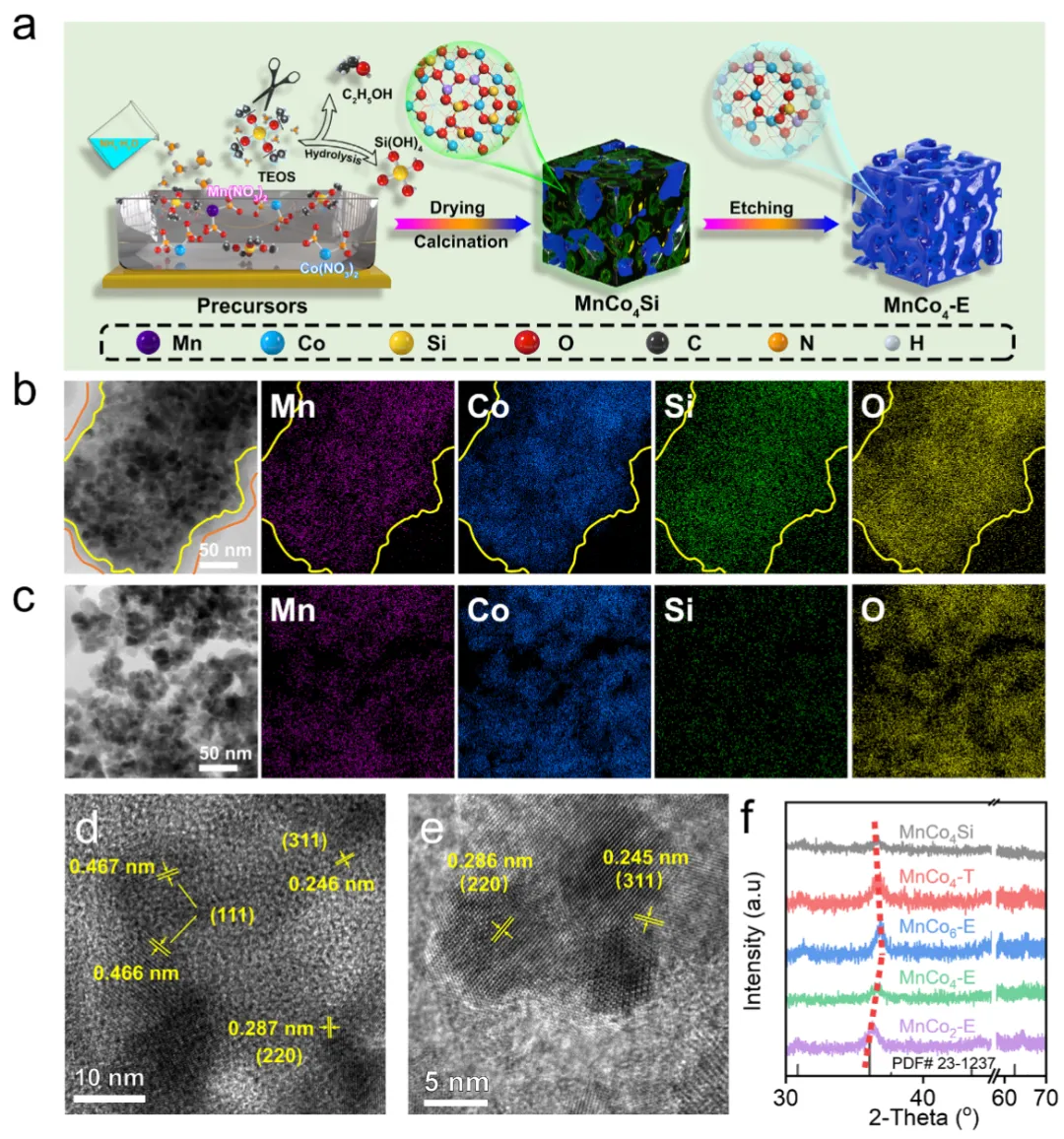
VOCs是工业源主要污染物之一,催化氧化法虽能将其彻底转化为CO₂和H₂O,但VOCs分子中稳定的C=O、C-C、C-H键往往需要较高温度才能断裂。问题的关键在于催化剂能否在低温下高效活化氧气——包括气相分子氧和表面晶格氧。尖晶石氧化物(AB₂O₄)因其结构可调、电子灵活而备受关注,但传统策略多依赖构筑氧空位,从分子轨道层面调控金属-氧共价性来协同活化两类氧物种的机理尚不清晰,成为制约低温性能的核心科学难题。2026年4月10日,南昌大学彭洪根、纪建团队在《Advanced Science》期刊发表题为“Reinforcing Oxygen Activation of Spinel Oxide via Mn─O Covalency Engineering for VOCs Oxidation”的研究论文。本研究提出通过削弱Mn-O共价性来增强氧活化的新思路。团队采用原位硬模板法向MnCo尖晶石中引入硅,诱导Mn-O键伸长、电荷重新分布,形成Mn⁴⁺-O-Co³⁺活性中心,成功实现了分子氧(生成超氧/过氧物种)与晶格氧(加速氧空位循环)的双重活化。最优催化剂MnCo₄-E对乙酸乙酯、甲苯、丙烷的T₉₀分别低至168、226、260°C,且抗水性优异、100小时不失活。结合原位光谱与DFT计算,研究揭示了弱化Mn-O共价性可降低乙酸盐氧化的决速步能垒,并将该策略验证于MnOₓ、Co₃O₄、MnCeOₓ等多种体系,展现出良好普适性。图1 催化剂合成与结构表征图1展示了通过原位硬模板法合成MnCo₄-E的全过程。碱蚀刻去除大部分硅后,MnCo₄-E形成了开放的多孔结构(图S1),TEM元素面分布(图1b-c)显示Mn、Co均匀分散,未发生相偏析。HRTEM(图1d-e)测得(311)晶面间距从0.242 nm增至0.245 nm,XRD(图1f)中衍射峰左移,共同证实了硅掺入引起的晶格膨胀。N₂吸附-脱附(图S4)表明,碱蚀刻后比表面积从68.6提升至81.8 m²·g⁻¹,为反应物提供了更丰富的可及活性位点。
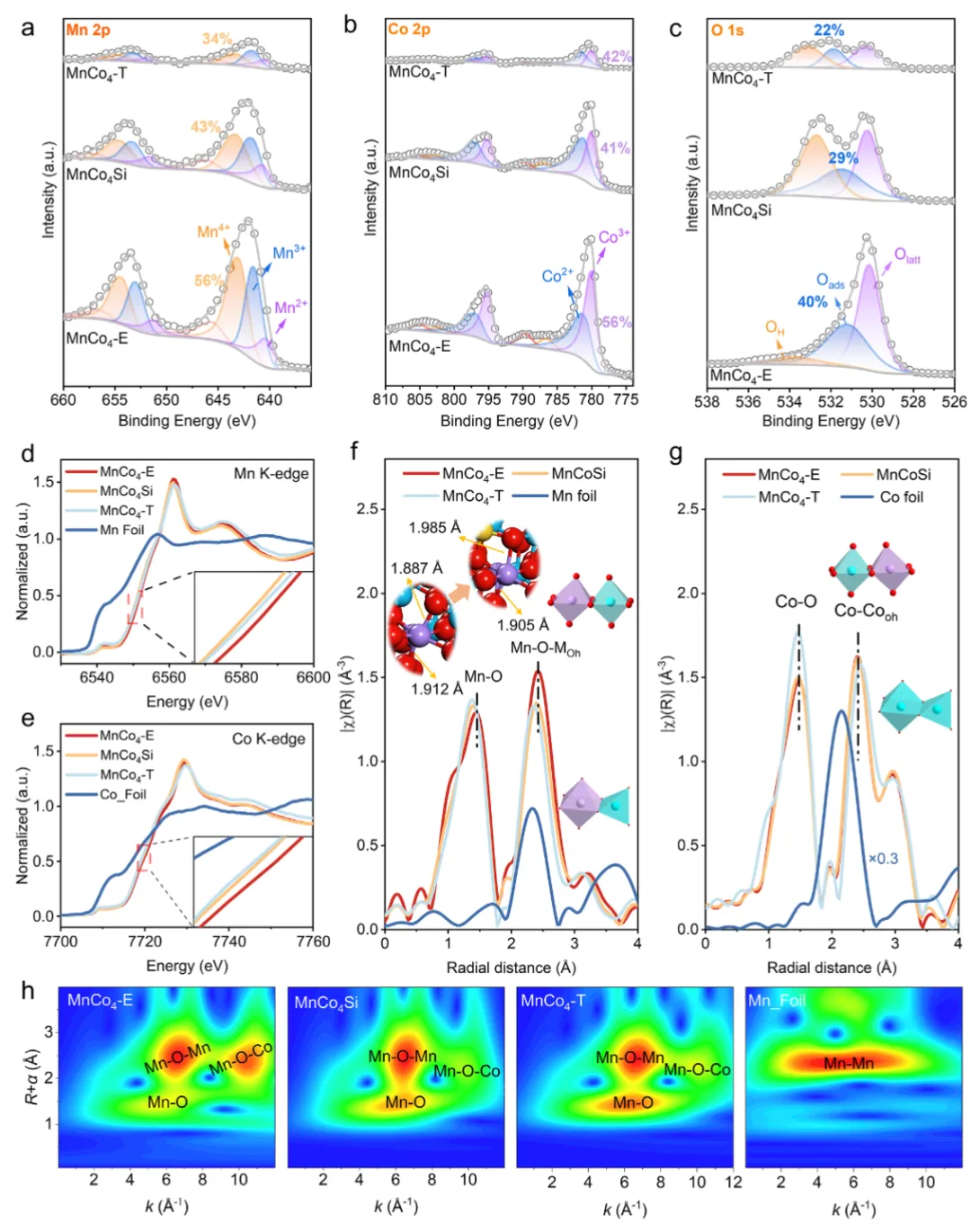
图2 催化剂的电子结构与配位环境XPS分析(图2a-c)显示,MnCo₄-E表面Mn⁴⁺/Co³⁺比例最高,吸附氧占比(0.40)远高于MnCo₄-T(0.22),且Mn 2p结合能正移、O 1s负移,表明电子从Mn向O转移,Mn-O共价性被削弱。XANES(图2d-e)证实Mn和Co平均氧化态升高。EXAFS(图2f-g)中Mn-O键长从1.91 Å伸长至1.93 Å,结合DFT计算的电荷密度差(图S16),从原子尺度确认了硅掺入导致Mn-O键弱化。Raman光谱(图S17)中Mn-O振动峰红移、力常数降低,进一步佐证。小波变换(图2h)清晰分离了Mn-O、Mn-O-Mn和Mn-O-Co散射信号,验证了Mn与Co的原子级混合。
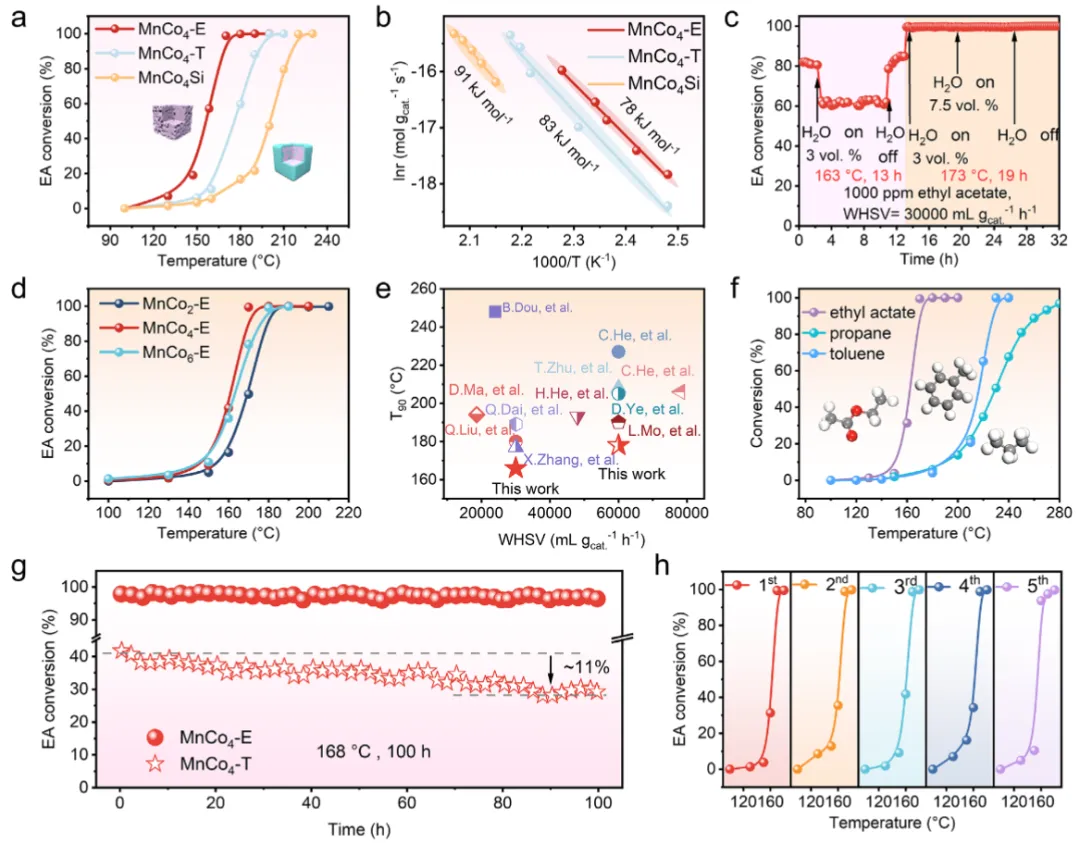
图3 催化性能评估图3证实了MnCo₄-E优异的催化性能。在EA氧化中,其T₉₀低至168°C,而对比样MnCo₄-T在170°C时转化率不足40%(图3a)。表观活化能仅78 kJ·mol⁻¹(图3b),低于MnCo₄-T(83)和MnCo₄Si(91),本征活性显著提升。即使在7.5%水蒸气、173°C条件下,活性仍保持稳定(图3c),展现出卓越的抗水干扰能力。通过调节Mn/Co比发现1:4为最佳(图3d)。与已报道的多种贵金属及过渡金属催化剂相比,MnCo₄-E活性处于领先水平(图3e)。此外,其对甲苯(T₉₀=226°C)和丙烷(T₉₀=260°C)同样高效(图3f),并在100小时连续运行及多次循环中性能几乎无衰减(图3g-h),实际应用潜力巨大。
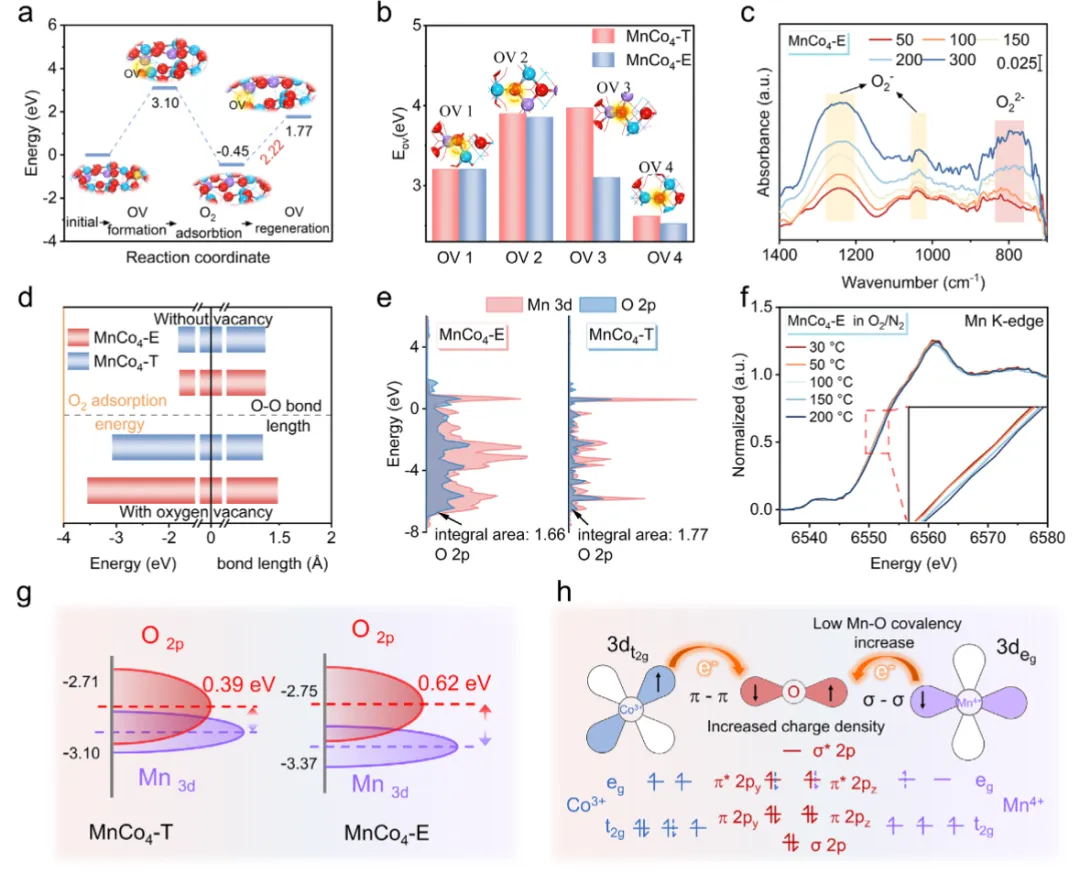
图4 氧活化机理探究图4从理论和实验两方面揭示了弱化Mn-O共价性增强氧活化的本质。DFT计算(图4a)表明,MnCo₄-E中氧空位形成能(3.10 eV)在O₂吸附后降至2.22 eV,说明其具备快速再生氧空位的能力。Co配位高的位点(OV₄)空位形成能最低(图4b),解释了高Co含量利于低温活性的现象。原位DRIFTS(图4c)在MnCo₄-E上检测到明显的超氧(O₂⁻)和过氧(O₂²⁻)物种信号,而MnCo₄-T仅生成少量O₂⁻,证实其更强的O₂活化能力。DFT进一步显示(图4d),MnCo₄-E上O₂吸附后O-O键显著伸长(1.23→1.49 Å),吸附能高达-3.55 eV。PDOS分析(图4e)表明Mn 3d与O 2p轨道杂化减弱,Mn-O共价性降低,有利于电子向O₂的π*轨道转移。原位XANES(图4f)中Mn边随温度升高向高能移动,证实了晶格氧的消耗与再补。图4h总结了电子从Mn经O向O₂传递、生成活性氧物种并参与氧化还原循环的完整机理。
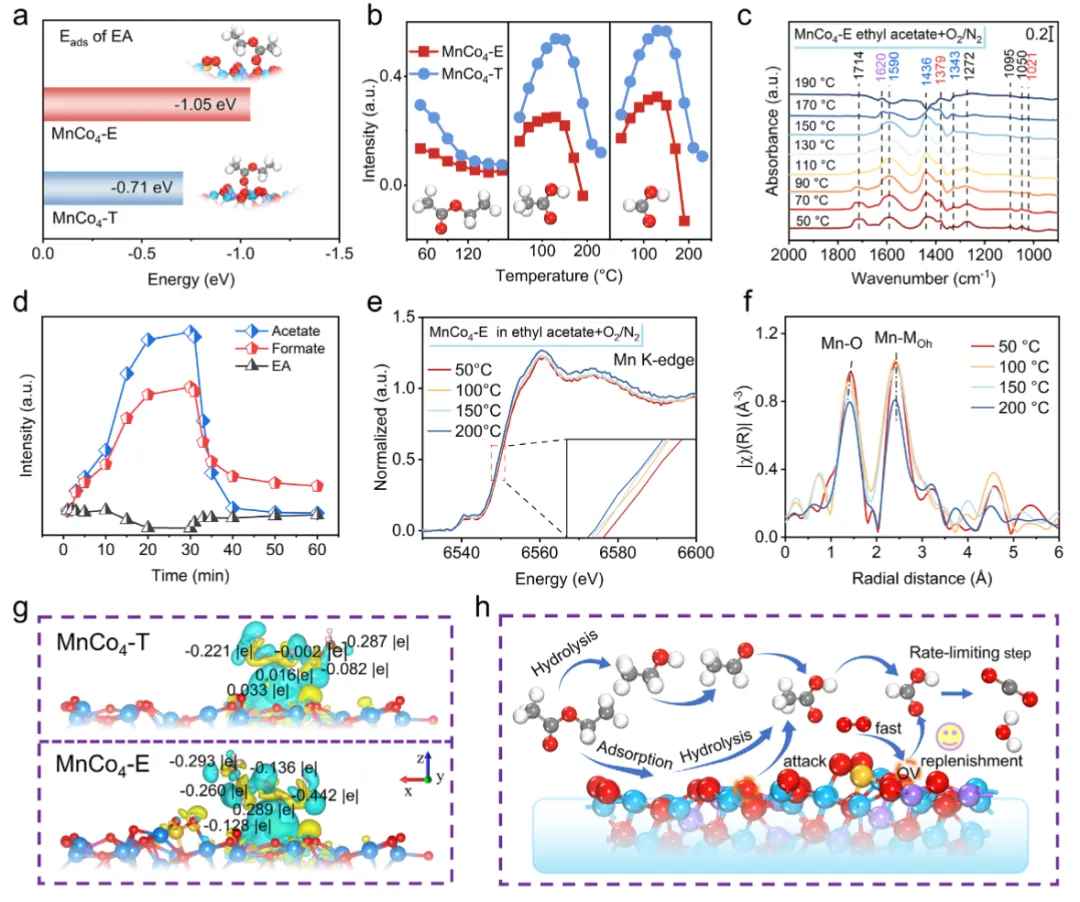
图5 乙酸乙酯氧化反应路径图5阐明了EA在MnCo₄-E上的降解路径与决速步。原位DRIFTS(图5b-c)显示,EA在室温下迅速水解为乙酸盐和甲酸盐,并在130°C以上开始被消耗。与MnCo₄-T相比,MnCo₄-E上乙酸盐/甲酸盐的转化温度更低(140°C vs 170°C),证明弱化Mn-O共价性有效降低了决速步(羧酸盐氧化)的能垒。气氛切换实验(图5d)表明,无氧条件下乙酸盐积累并生成乙烯副产物,通入O₂后迅速完全氧化,证实了反应遵循Mars-van Krevelen(MVK)机理。原位XAFS(图5e-f)显示,反应中Mn氧化态降低、Mn-O配位数下降,直接证明了晶格氧的参与。电荷密度差(图5g)表明,MnCo₄-E向EA转移更多电子,使其α-碳更具电负性,易受晶格氧攻击。图5h总结了EA先水解为乙酸盐、再经甲酸盐路径最终矿化为CO₂和H₂O的完整反应路径。
总之,本研究提出并验证了一种通过削弱Mn-O共价性来增强尖晶石氧化物低温氧活化能力的新策略。其核心机制在于:原位硬模板法掺入硅,诱导Mn-O键伸长与电荷重新分布,削弱共价性,从而促进电子从Mn向O转移,降低氧气解离能垒,实现分子氧(生成O₂⁻/O₂²⁻)与晶格氧(加速氧空位循环)的双重活化,并显著加速了乙酸盐氧化的决速步骤。催化性能方面,最优催化剂MnCo₄-E对乙酸乙酯、甲苯和丙烷均表现出低T₉₀、高反应速率、低表观活化能,以及优异的耐水性和超过100小时的长期稳定性。应用价值在于,该策略不仅为设计高效、廉价的VOCs氧化催化剂提供了分子层面的新思路,而且成功拓展至MnOₓ、Co₃O₄等体系,展示了良好的普适性。当然,该前沿研究仍有待进一步深入,例如在更复杂的混合VOCs气氛下的性能、大尺寸催化剂制备以及工业化反应器设计等挑战。但其在实现低能耗、高效率的VOCs治理方面展现出的巨大潜力,无疑值得环境催化产业界的高度期待。本研究提出并验证了一种“以废治废”的新策略:采用闪速焦耳热技术将废弃轮胎碎片快速转化为铁负载碳基吸附剂,用于高效去除水中聚苯乙烯纳米塑料。该策略的核心在于FJH的超快热循环(毫秒级升温至>2000 K,200 K/s冷却)不仅实现了碳前驱体的石墨烯化,还促进了铁纳米颗粒在碳层间的均匀分散,避免了传统热解方法中易出现的金属团聚问题。机制上,吸附过程由静电吸引、疏水作用、π-π堆积与Fe介导表面络合四种效应协同驱动,使其在宽pH范围(3–10)及实际水体中仍保持高效性能。性能上,最优材料FJH-0.5对PSNPs的去除率达99.4%,最大吸附容量接近1000 mg/g,远超多数文献报道值,且具有超顺磁性(14.68 emu/g)便于磁分离。应用价值方面,废吸附剂可通过二次FJH处理快速再生,三次循环后去除率仍高于94.2%,结合废轮胎的低成本与FJH的低能耗(对比传统热解),展现出良好的工程应用潜力。未来可进一步探索该策略对其他类型纳米塑料、微塑料乃至新兴有机污染物的去除能力,以及FJH参数对材料结构与性能的精细调控机制。
本研究提出并验证了一种“以废治废”的新策略:采用闪速焦耳热技术将废弃轮胎碎片快速转化为铁负载碳基吸附剂,用于高效去除水中聚苯乙烯纳米塑料。该策略的核心在于FJH的超快热循环(毫秒级升温至>2000 K,200 K/s冷却)不仅实现了碳前驱体的石墨烯化,还促进了铁纳米颗粒在碳层间的均匀分散,避免了传统热解方法中易出现的金属团聚问题。机制上,吸附过程由静电吸引、疏水作用、π-π堆积与Fe介导表面络合四种效应协同驱动,使其在宽pH范围(3–10)及实际水体中仍保持高效性能。性能上,最优材料FJH-0.5对PSNPs的去除率达99.4%,最大吸附容量接近1000 mg/g,远超多数文献报道值,且具有超顺磁性(14.68 emu/g)便于磁分离。应用价值方面,废吸附剂可通过二次FJH处理快速再生,三次循环后去除率仍高于94.2%,结合废轮胎的低成本与FJH的低能耗(对比传统热解),展现出良好的工程应用潜力。未来可进一步探索该策略对其他类型纳米塑料、微塑料乃至新兴有机污染物的去除能力,以及FJH参数对材料结构与性能的精细调控机制。
本研究提出并验证了一种“以废治废”的新策略:采用闪速焦耳热技术将废弃轮胎碎片快速转化为铁负载碳基吸附剂,用于高效去除水中聚苯乙烯纳米塑料。该策略的核心在于FJH的超快热循环(毫秒级升温至>2000 K,200 K/s冷却)不仅实现了碳前驱体的石墨烯化,还促进了铁纳米颗粒在碳层间的均匀分散,避免了传统热解方法中易出现的金属团聚问题。机制上,吸附过程由静电吸引、疏水作用、π-π堆积与Fe介导表面络合四种效应协同驱动,使其在宽pH范围(3–10)及实际水体中仍保持高效性能。性能上,最优材料FJH-0.5对PSNPs的去除率达99.4%,最大吸附容量接近1000 mg/g,远超多数文献报道值,且具有超顺磁性(14.68 emu/g)便于磁分离。应用价值方面,废吸附剂可通过二次FJH处理快速再生,三次循环后去除率仍高于94.2%,结合废轮胎的低成本与FJH的低能耗(对比传统热解),展现出良好的工程应用潜力。未来可进一步探索该策略对其他类型纳米塑料、微塑料乃至新兴有机污染物的去除能力,以及FJH参数对材料结构与性能的精细调控机制。
中科精研自主研发的焦耳加热设备,采用先进的毫秒级超快升温技术,可在极短时间内实现高温处理,广泛应用于纳米材料制备、催化剂合成、电池材料研发等领域。毫秒级升温速率 | 精准温控系统 | 多种气氛环境 | 操作简便安全