Angew | 南昌大学【徐香兰】/【王翔】Angew: 亲氧Ru-O-Mn²⁺界面CO₂甲烷化低温超高活性!邓绍荣一作!
亲爱的读者们,不星标《纵横科研》公众号,会收不到我们的最新推送点击公众号主页右上角,星标《纵横科研》,不错过每一条科研资讯Oxyphilic Ru-O-Mn2+ Interfaces on Ru/MnO Catalysts: Unprecedented Activity for Low-Temperature CO2 Methanation
https://doi.org/10.1002/anie.6087744利用可再生氢气催化二氧化碳甲烷化是实现碳中和与可再生能源存储的前景广阔的策略,但在低温下实现高效转化仍面临巨大挑战。本研究报道了一种具有原位构建的高亲氧性界面的催化剂,其表现出卓越的二氧化碳甲烷化性能。在180°C和36,000 mL g⁻¹ h⁻¹空速条件下,该催化剂实现了94.9%的二氧化碳转化率,其甲烷产率超越了当前最先进的催化剂,并保持了优异的稳定性。结合实验与理论研究表明,该亲氧界面是关键活性中心,揭示了界面亲氧物种数量、弱吸附二氧化碳量与催化转换频率之间的直接正相关关系。该亲氧界面位点建立了广泛的弱吸附二氧化碳网络,从而促进了低温下的二氧化碳活化。该策略成功拓展至镍基体系,印证了二氧化锰介导界面工程的普适性。这些发现为低温催化确立了新的设计范式,并深化了对多相催化中界面亲氧性的基础理解。
全球气候危机正从根本上改变地球生态系统,其主要驱动力是大气中二氧化碳浓度已超出工业革命前水平的150%。这一紧迫形势使得催化二氧化碳转化为高价值燃料和化学品,成为实现碳循环闭合的前沿研究重点。其中,二氧化碳甲烷化极具前景,其产物甲烷是一种碳中性能源载体,与现有天然气基础设施兼容,可为可再生能源存储和电网平衡提供实用的电转气解决方案,并在深空探测中展现出新兴应用前景。二氧化碳甲烷化是放热反应,而逆水煤气变换副反应是吸热反应,热力学上低温有利于甲烷生成。然而,低温下该反应面临显著的动力学限制,需要高效催化剂以实现最佳反应速率。因此,开发能够在低温下实现工业可行速率的高级催化剂,成为一个关键的材料设计挑战。
在中等温度下,二氧化碳在贵金属纳米颗粒上解离为一氧化碳中间体是明确的反应过程,其中钌纳米颗粒因其最优的一氧化碳解离吸附能,在二氧化碳甲烷化中表现出卓越的活性和选择性。因此,一氧化碳中间体是大多数钌基体系中的关键中间体。然而,一个基本问题仍然存在:二氧化碳解离为一氧化碳中间体与随后的甲烷化反应,何者为速率控制步骤,这一区分对于理性设计高效低温催化剂至关重要。大多数文献认为,由于二氧化碳分子中碳氧键的化学惰性,其活化和解离是关键步骤。一些研究也指出,钌催化剂上二氧化碳甲烷化的动力学能垒,与在钌表面形成接近饱和吸附的一氧化碳中间体及其随后的氢化有关,因为一氧化碳中的碳氧键比二氧化碳中的更强。先前研究表明,一氧化碳在钌上的甲烷化受钌颗粒尺寸和电子态影响,其中尺寸是主导因素。小尺寸钌颗粒表现出高活性的一氧化碳甲烷化能力。在这种情况下,二氧化碳解离为一氧化碳中间体实际上限制了整体反应速率,成为必须考虑的关键因素。优化金属颗粒及其与载体的界面日益受到关注。金属‑载体界面因其独特的电子结构,在二氧化碳活化中起着关键作用,在特定界面位点可获得极高的活性。通过定制复杂的界面结构以获得高效的低温二氧化碳甲烷化催化剂,是一个有吸引力但极具挑战性的课题。
锰氧化物因其多价态性质,被广泛应用于催化氧化反应。然而,其在二氧化碳甲烷化中的应用仍显不足。这源于高价态锰氧化物在预还原或催化过程中会被还原为氧化锰。据报道,预还原的钌/氧化锰具有显著的亲氧性,即使在室温下暴露于空气中,氧化锰也会发生氧化和价态变化,这一现象在其他钌基体系中未被观察到。值得注意的是,研究者将金属对二氧化碳活化/解离的能力与亲氧性相关联,提出二氧化碳解离更易在高氧亲和力的金属上发生。此外,研究表明钌在氧化锰上高度分散,1 wt.% 钌/氧化锰中钌颗粒平均尺寸仅为1.6纳米。这些发现证明了氧化锰作为高效钌载体的巨大潜力。
利用氧空位促进二氧化碳吸附和解离,已被广泛报道为提升低温二氧化碳甲烷化性能的有效策略。在本工作中,我们提出了一种新颖的替代方案,即通过无煅烧的沉积‑沉淀法制备钌/氧化锰催化剂。值得注意的是,该催化剂表现出前所未有的低温活性,实现了创纪录的二氧化碳转化率和甲烷产率,其性能超越了所有当前最先进的类似催化剂。通过详细表征和密度泛函理论计算,我们阐明该反应由高亲氧性的钌‑氧‑锰界面驱动。至关重要的是,这种界面机制被证明从根本上优于传统的氧空位介导路径,为设计低温甲烷化催化剂提供了新的概念框架。
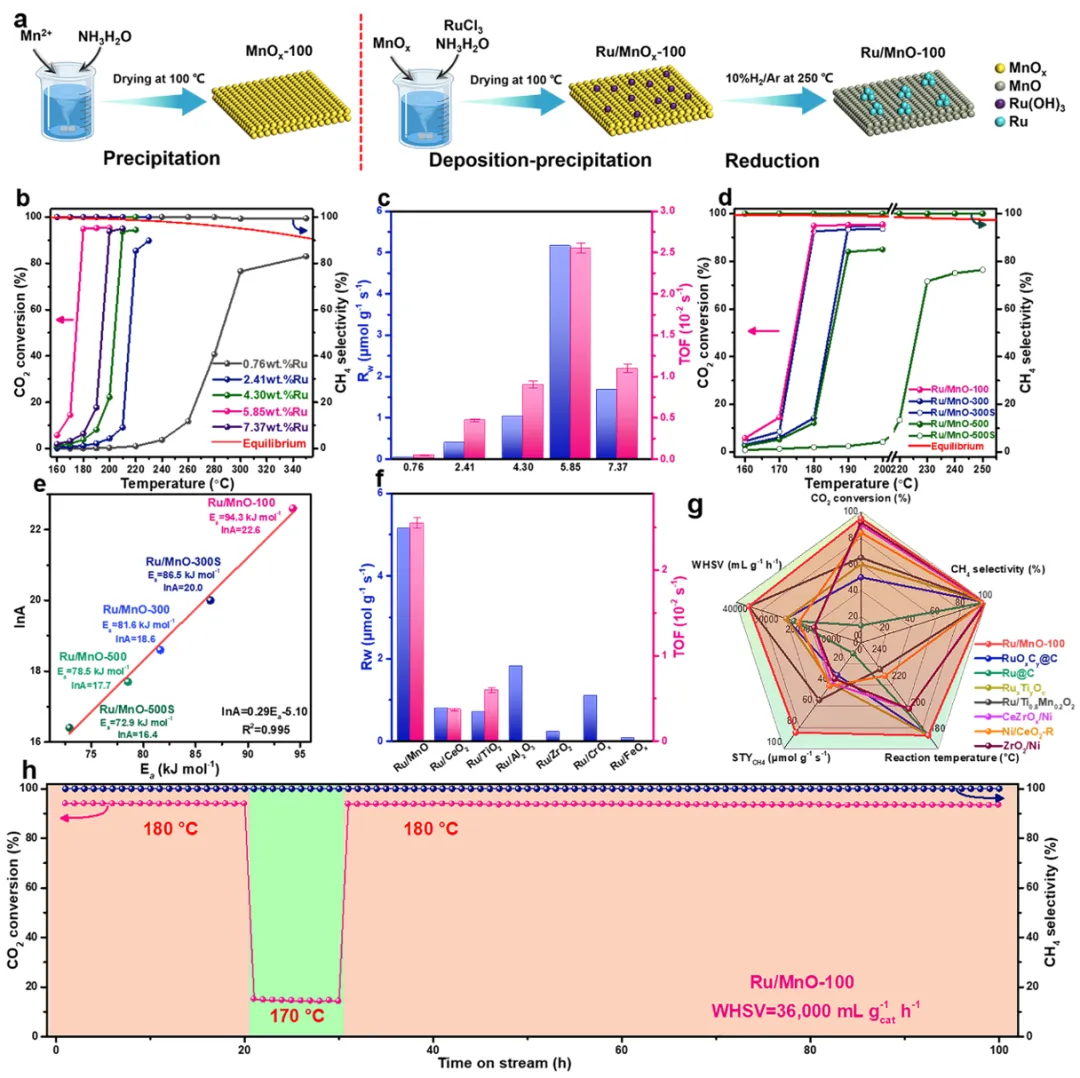
图1
(a) 最佳催化剂的合成流程;(b) 不同钌负载量催化剂的二氧化碳转化率和甲烷选择性;(c) 质量归一化反应速率与转换频率;(d) 不同处理条件下的催化剂性能与表观活化能、指前因子的关系;(e) 表观活化能对指前因子的依赖关系;(f) 不同载体上钌催化剂的活性对比;(g) 催化剂在五项关键指标上与前沿催化剂的对比;(h) 催化剂的稳定性测试。
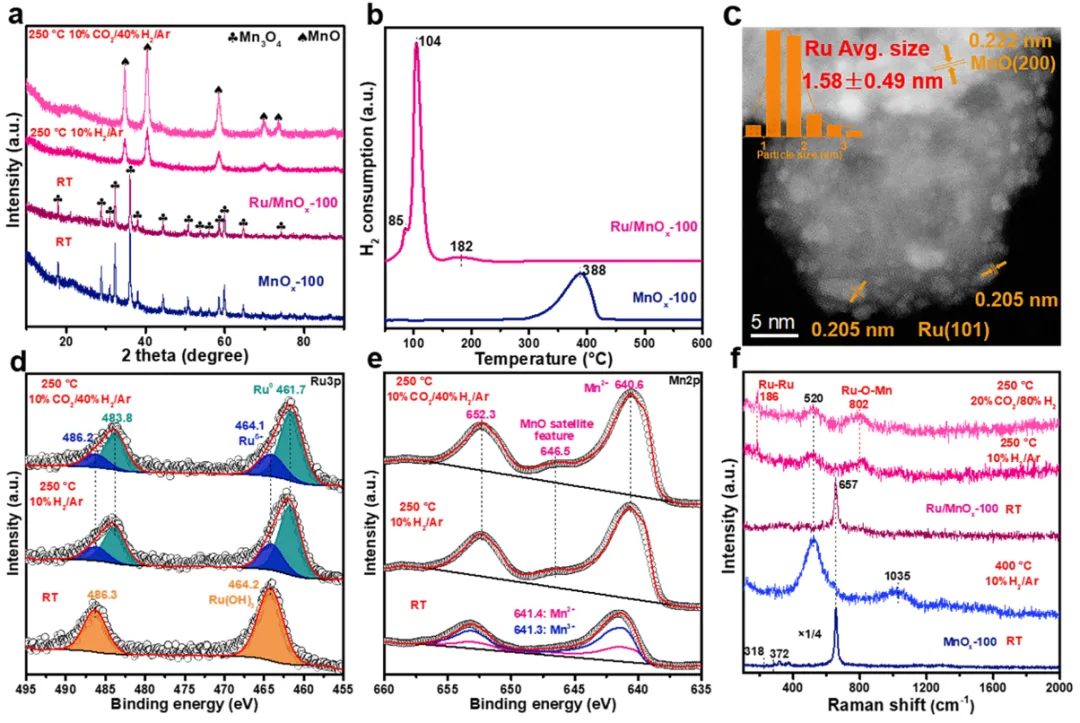
图2
(a) 载体和催化剂的常压原位X射线衍射谱;(b) 氢气程序升温还原谱;(c) 高角环形暗场扫描透射电子显微镜图像与钌平均粒径统计;(d-e) 在不同条件下催化剂的准原位X射线光电子能谱;(f) 载体和催化剂的原位拉曼光谱。
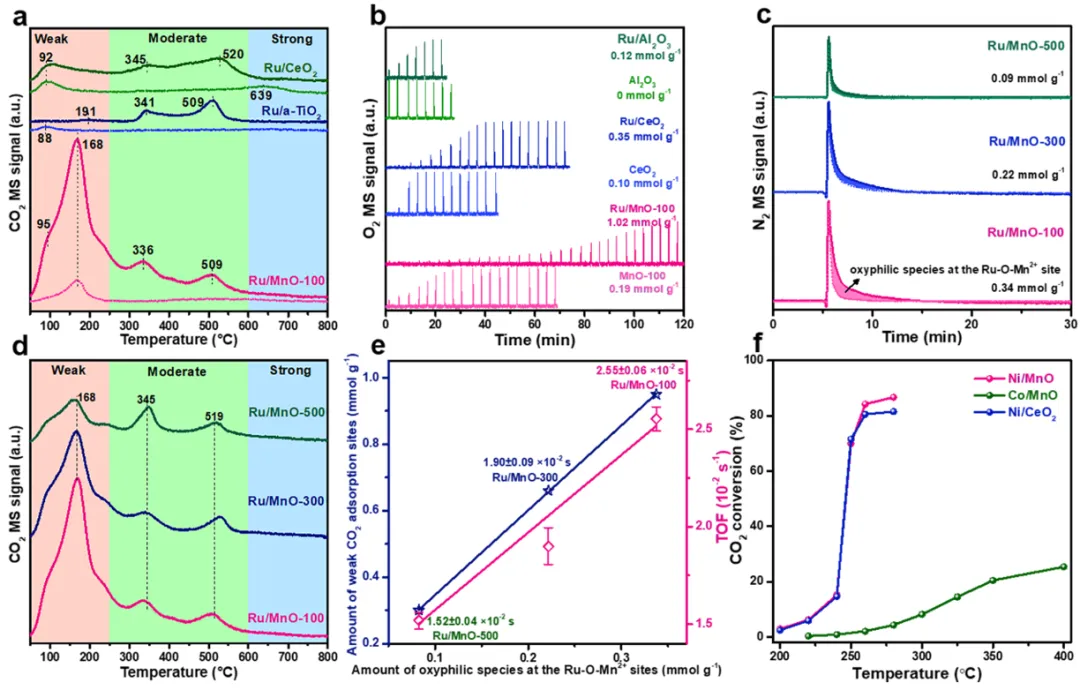
图3
(a) 催化剂和相应载体的二氧化碳程序升温脱附谱;(b) 催化剂和相应载体的氧气脉冲吸附谱;(c) 通过一氧化二氮滴定结合差分氮气信号对催化剂界面亲氧物种的定量;(d) 催化剂的二氧化碳程序升温脱附谱;(e) 界面亲氧物种数量、弱吸附二氧化碳位点数量与转换频率的关联;(f) 不同催化剂的二氧化碳甲烷化转化率曲线。
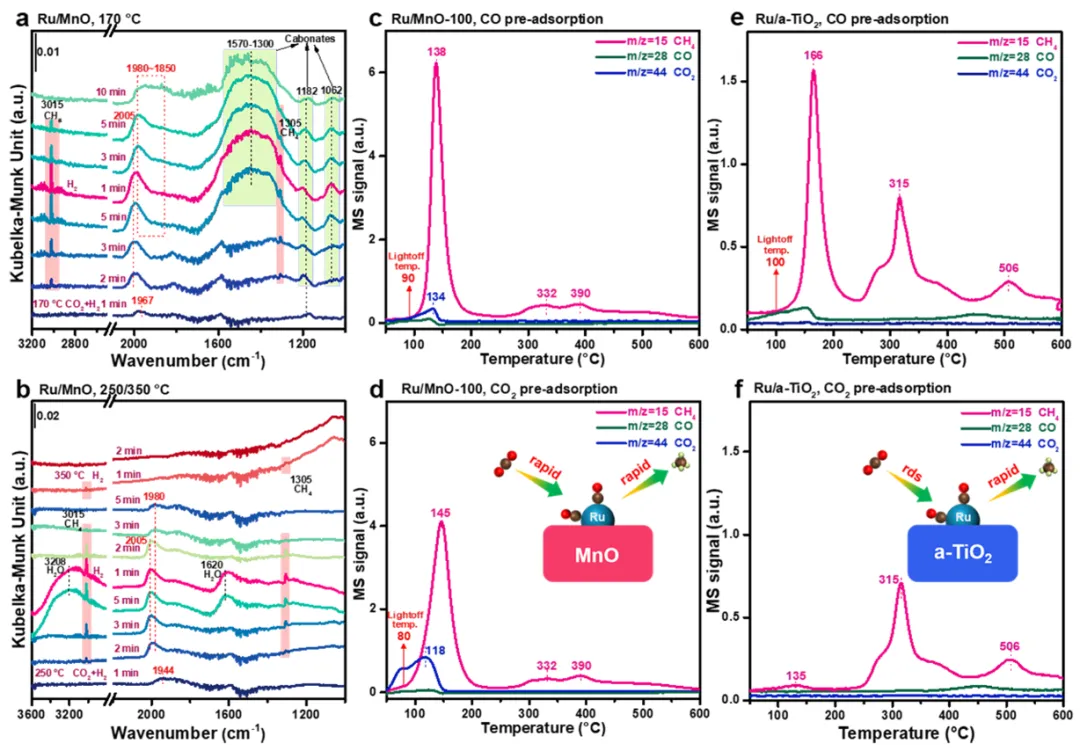
图4
在二氧化碳/氢气混合气中反应,随后切换为氢气/氩气的条件下,催化剂的原位漫反射红外傅里叶变换光谱:(a) 170°C与(b) 250/350°C下;(c-d) 以催化剂和(e-f) 另一种催化剂为对象,在经一氧化碳/氦气或纯二氧化碳气流预处理后,在氢气/氩气流中的程序升温表面反应‑质谱谱图。
图5
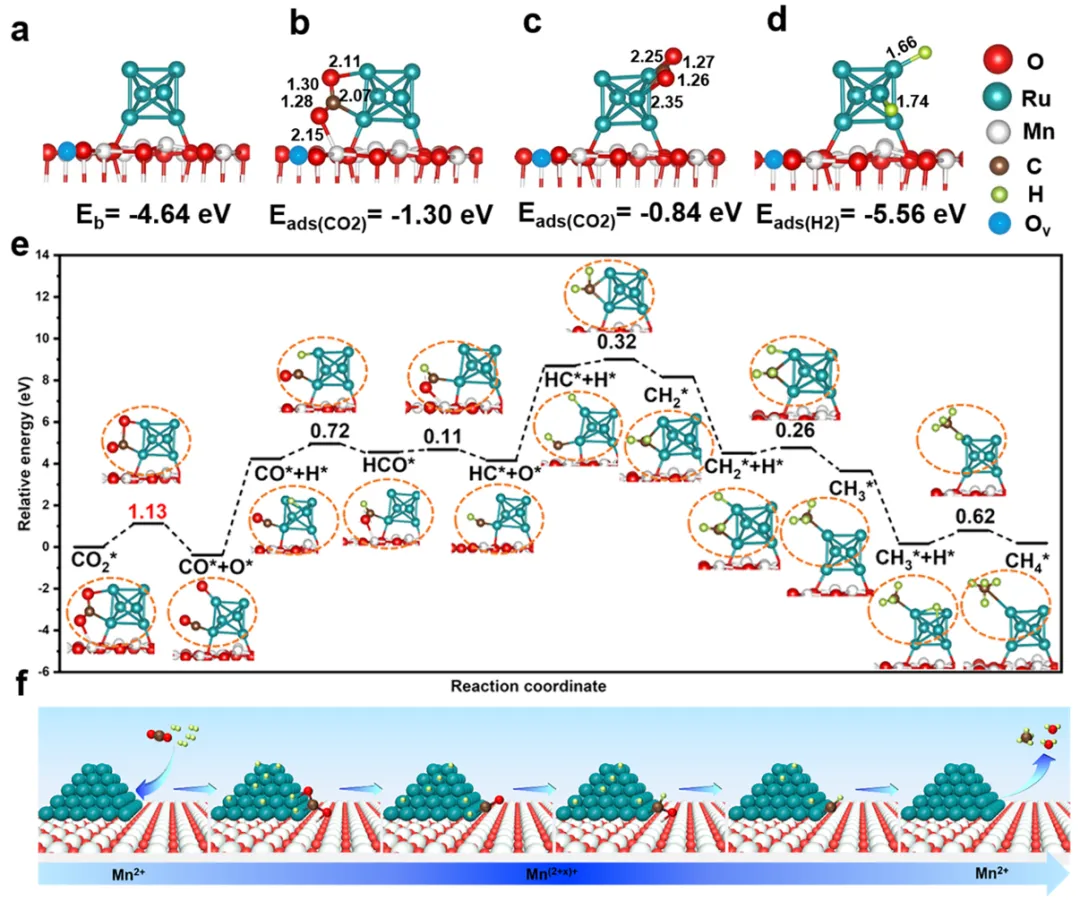
(a) 负载钯簇模型的结合能;(b-c) 二氧化碳在界面位点上吸附的结构模型;(d) 氢气吸附结构模型;(e) 二氧化碳在模型上的最优甲烷化路径及各步骤活化能;(f) 亲氧性界面对二氧化碳甲烷化的关键作用示意图。
总之,我们开发了一种简便的无煅烧沉积‑沉淀策略,合成了一种在低温二氧化碳甲烷化中表现出空前效率的钌/氧化锰催化剂。该催化剂在180°C、常压和高空速条件下,实现了94.9%的二氧化碳转化率和84.7微摩尔每克每秒的甲烷产率,确立了新的性能标杆,超越了当前最先进的同类催化剂,并保持了优异的稳定性。结合实验与理论研究,我们确定亲氧性的钌‑氧‑锰界面而非氧空位是决定性活性中心。研究表明,界面亲氧物种的数量严格线性决定了弱吸附二氧化碳的能力,进而决定了催化转换频率。从机理上看,这些高亲氧性的界面位点促进了二氧化碳以弱吸附状态被捕获,从而降低了其解离为一氧化碳中间体的能垒。重要的是,该策略成功拓展至镍/氧化锰体系,印证了二氧化锰介导界面工程的普适性。本工作不仅为甲烷化提供了高性能解决方案,也为设计亲氧性金属‑载体界面提供了基础性见解,对传统氧空位介导机制的主导地位提出了挑战。








 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
