本研究以多株肺癌患者类器官(LCO)为核心模型(图 1),开展 CRISPR 全基因组筛选。经过电离辐射处理后对比基因表达差异,最终锁定 HDAC4 为放疗耐药核心基因。实验验证:辐射会上调 HDAC4 表达,敲低该基因后类器官死亡比例显著上升;过表达野生型 HDAC 会强化耐药,而失活突变体无此效果,初步证实 HDAC4 依靠类泛素化活性驱动放疗抵抗,肺癌类器官为靶点筛选提供了高度贴合临床的可靠模型。
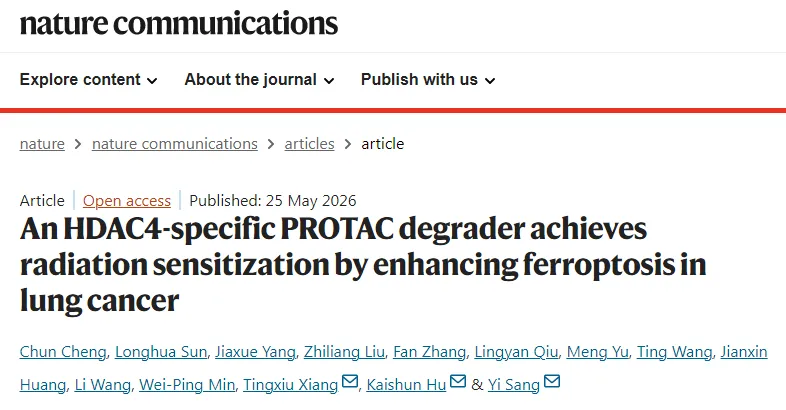
本图依托肺癌患者来源类器官(LCO)结合 CRISPR 文库筛选,定位介导肺癌放疗抵抗的关键基因。研究选用两株放疗表型差异显著的肺癌类器官,接受 9 Gy 电离辐射(IR)处理,SYTOX Green 染色结果显示,辐射后耐药型类器官死亡细胞占比远低于敏感株(图 1a、b)。基于全基因组 CRISPR 敲除文库,对辐射组与对照组进行基因差异分析,通过 β 值排序筛选出差异基因(图 1c、d),取两株类器官共同下调的前 20 个基因,交集结果明确 HDAC4 为核心候选靶点(图 1e)。免疫荧光证实,电离辐射会显著上调类器官内 HDAC4 蛋白表达(图 1f)。梯度辐射实验表明,辐射剂量越高,类器官存活能力越强、细胞死亡越少;敲低 HDAC4 后,不同辐射剂量下类器官存活率明显下降、死亡细胞增多(图 1g-j)。
细胞实验显示,敲除 HDAC4 会大幅提升脂质活性氧水平,触发典型铁死亡特征,电镜也观察到铁死亡标志性线粒体形态变化。使用铁死亡抑制剂处理后,该表型被逆转,明确 HDAC4 通过抑制铁死亡产生放疗抗性,厘清了下游核心细胞死亡通路。
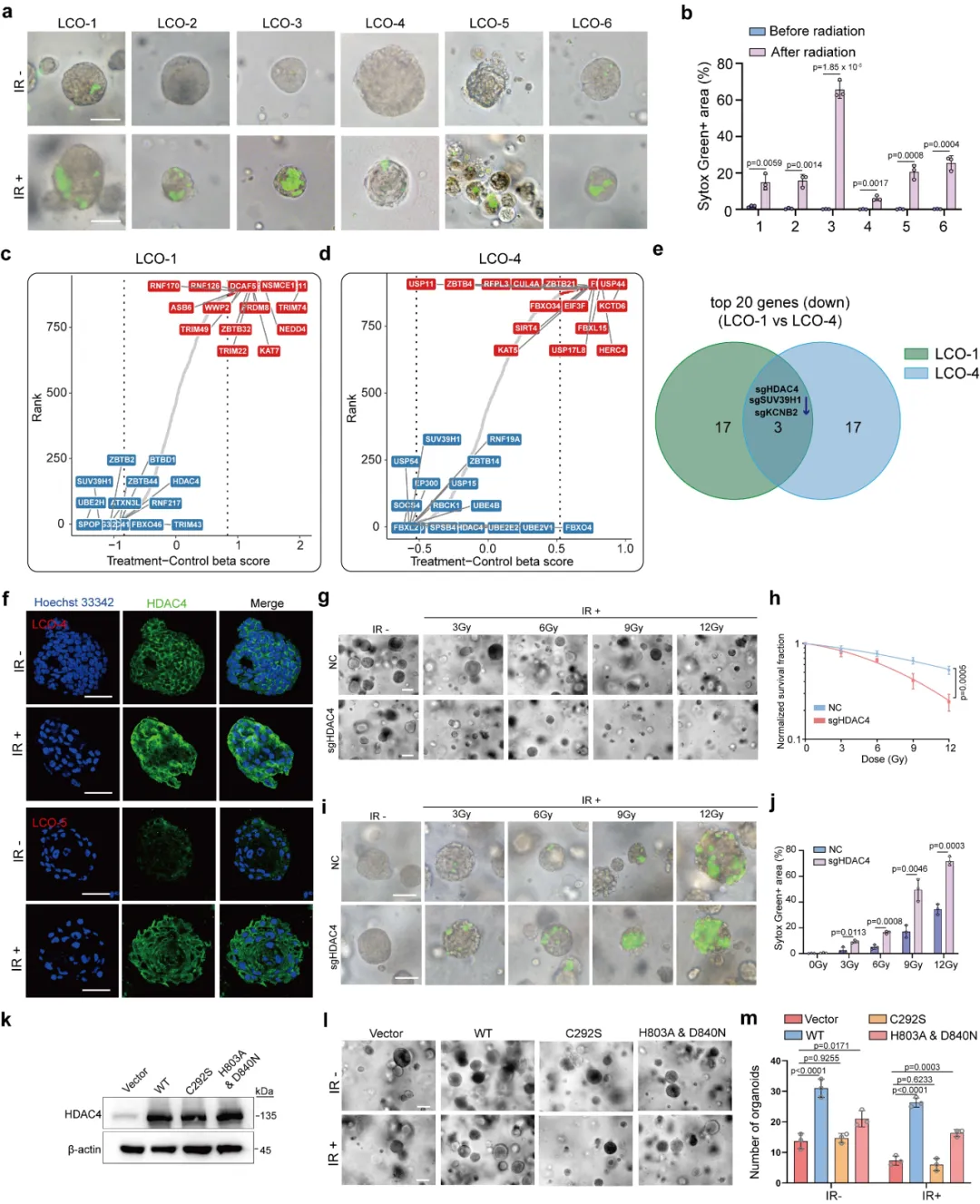
本图阐明 HDAC4 调控放疗抵抗的核心通路 —— 铁死亡。首先通过 shRNA 构建 HDAC4 敲低肺癌细胞系(图 2a),采用 C11-BODIPY 探针检测脂质活性氧(lipid ROS),这是铁死亡标志性指标。结果显示,辐射联合 HDAC4 敲低后,细胞内脂质 ROS 水平大幅升高(图 2b-e)。透射电镜观察到典型铁死亡形态:线粒体皱缩、膜密度增高、外膜破损(图 2f)。使用铁死亡特异性抑制剂 Ferrostatin-1(Fer-1)干预后,HDAC4 敲低引发的脂质 ROS 积累被显著逆转(图 2g-j),直接证明 HDAC 通过抑制铁死亡发挥作用。进一步构建 HDAC4 敲除细胞,并分别回补野生型、去乙酰化酶失活、SUMO 酶失活突变体(图 2k)。
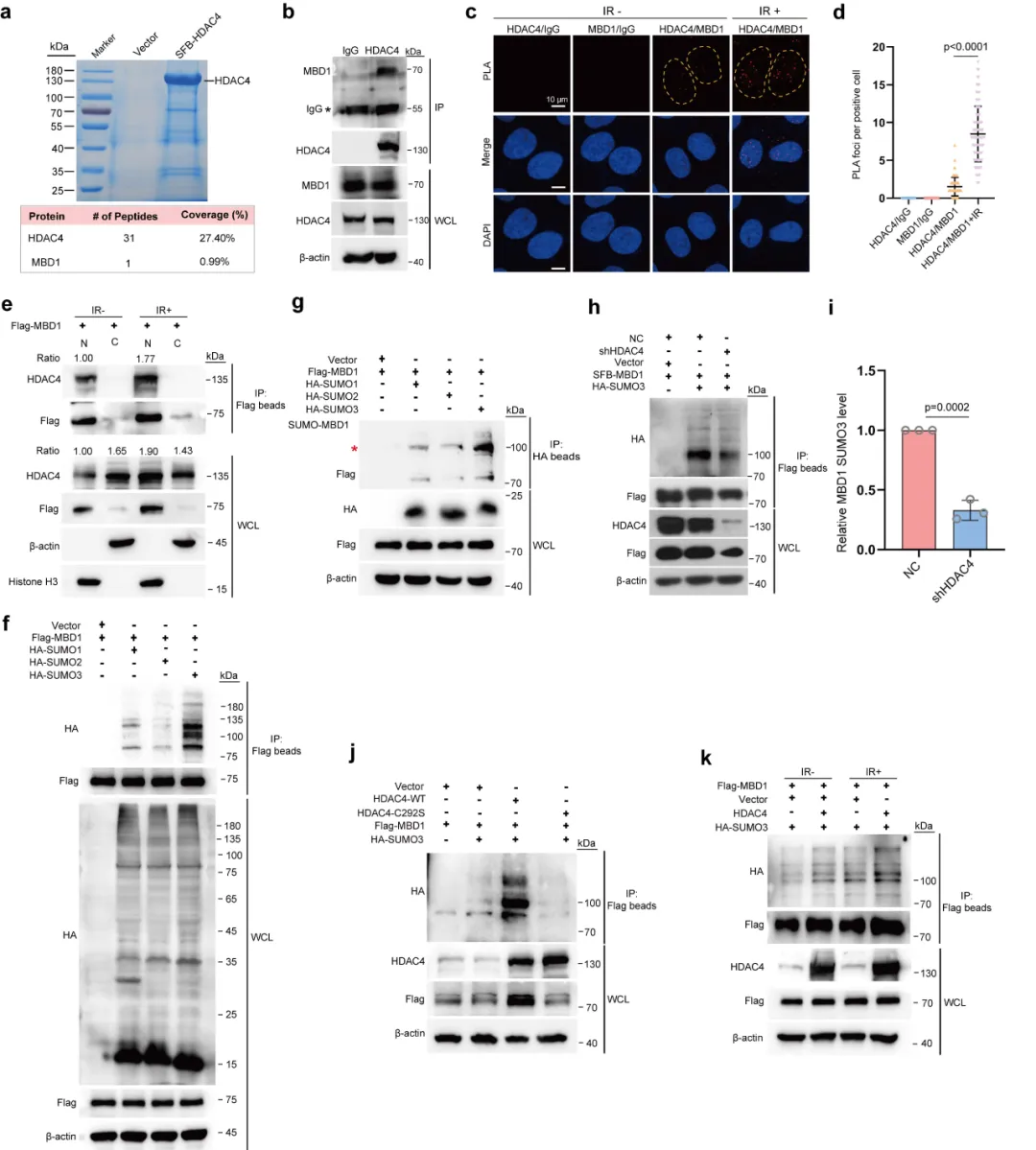
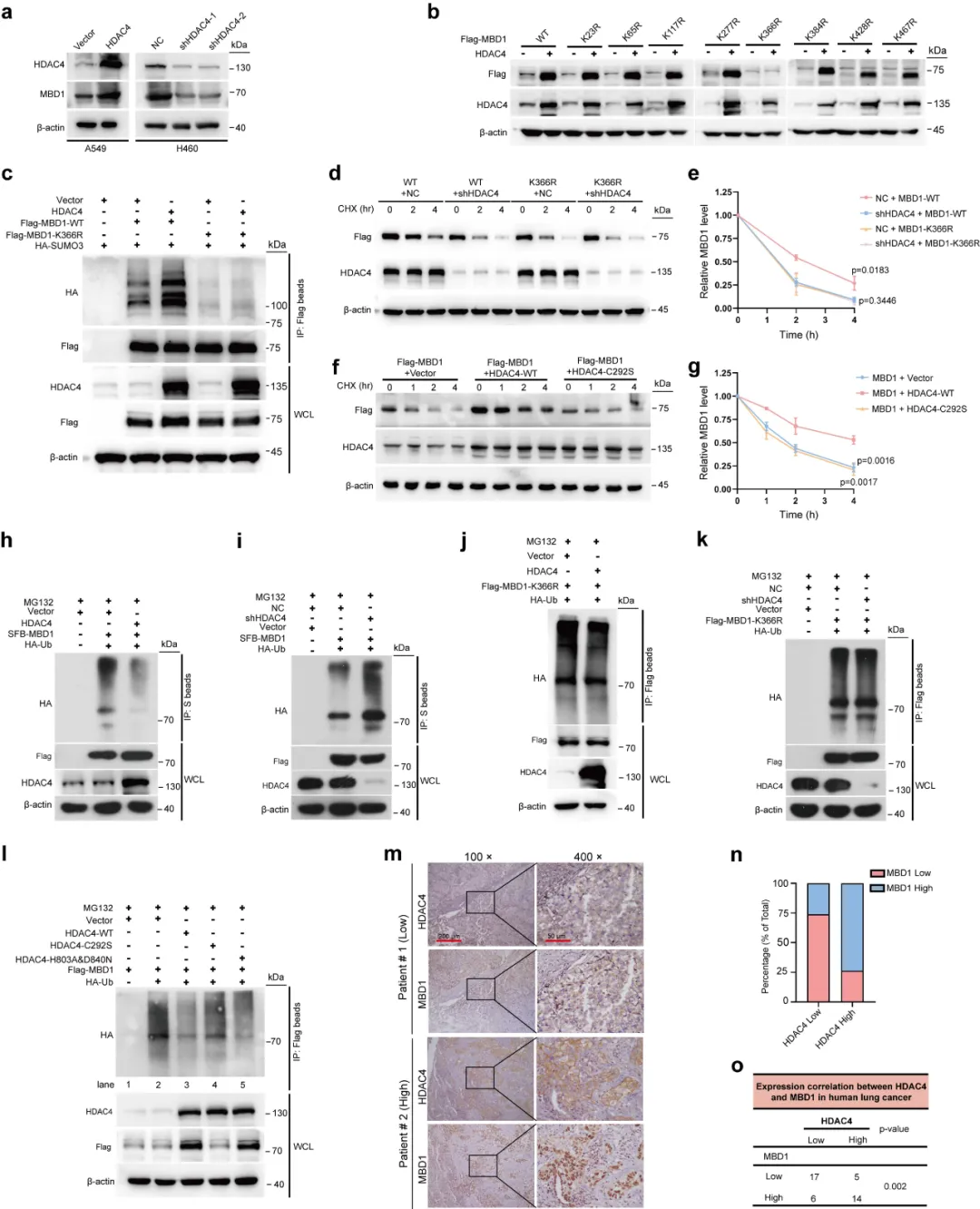
通过蛋白互作筛选找到下游靶点 MBD1(图 3),证实 HDAC4 可直接结合 MBD1 并催化其 SUMO3 类泛素化。该修饰发生在 K366 位点(图 4),能阻碍 MBD1 泛素化降解、延长蛋白半衰期,临床样本也验证二者表达呈正相关,完整阐明蛋白修饰级联反应。
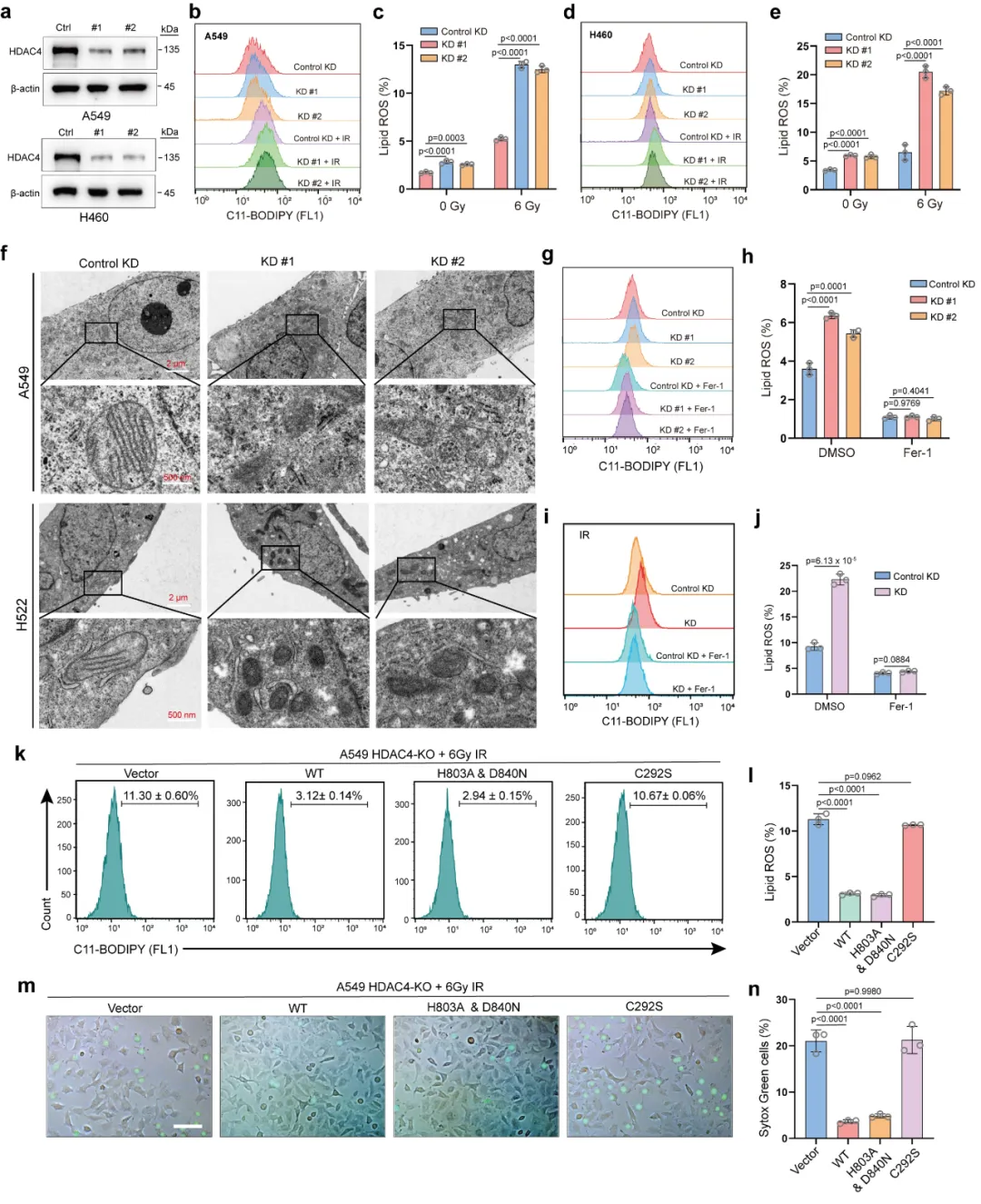
本图鉴定 HDAC4 的直接互作蛋白 MBD1,并验证辐射对二者结合的调控效应。首先通过串联亲和纯化联合质谱分析(图 3a),在 HDAC4 复合物中富集到甲基 CpG 结合蛋白 MBD1。内外源免疫共沉淀(Co-IP)实验(图 3b)反复验证,肺癌细胞中 HDAC4 与 MBD1 可稳定结合。邻近连接实验(PLA)直观显示,电离辐射处理后,细胞核内 HDAC4-MBD1 相互作用位点数量显著增加(图 3c、d),说明辐射会促进二者在核内结合。核质分离结合 Co-IP 实验(图 3e)进一步证实,辐射刺激下核内复合物含量明显上升。后续蛋白修饰实验表明,HDAC4 不影响 MBD1 乙酰化,但可特异性催化 MBD1 发生 SUMO3 修饰(图 3f-g);敲低 HDAC4 会降低 MBD1 的 SUMO3 水平,过表达野生型 HDAC 可上调修饰,而 SUMO 酶失活突变体无此功能(图 3h-j)。辐射处理也会进一步增强该 SUMO 化修饰(图 3k)。
本图聚焦 HDAC4 通过 SUMO 化调控 MBD1 蛋白稳定性的分子机制。实验发现,改变 HDAC4 表达仅影响 MBD1 蛋白量,不改变其 mRNA 水平,说明调控发生在蛋白层面(图 4a)。结合位点突变实验显示,MBD1 的 K366 位点是核心 SUMO 修饰位点,该位点突变后,HDAC4 无法再提升 MBD1 蛋白丰度(图 4b、c)。蛋白半衰期实验(图 4d-g)证实:HDAC4 敲低会缩短野生型 MBD1 半衰期,但对 K366 突变体无影响;HDAC4 的 SUMO 酶活性缺失后,也无法稳定 MBD1。泛素化实验揭示分子竞争关系:SUMO 修饰会占据 MBD1 的赖氨酸位点,阻碍泛素结合(图 4h-l)。HDAC4 高表达时,MBD1 泛素化水平降低、降解减少;K366 突变后该保护效应消失。最后临床样本免疫组化(图 4m-o)验证,肺癌组织中 HDAC 与 MBD1 蛋白表达呈显著正相关。
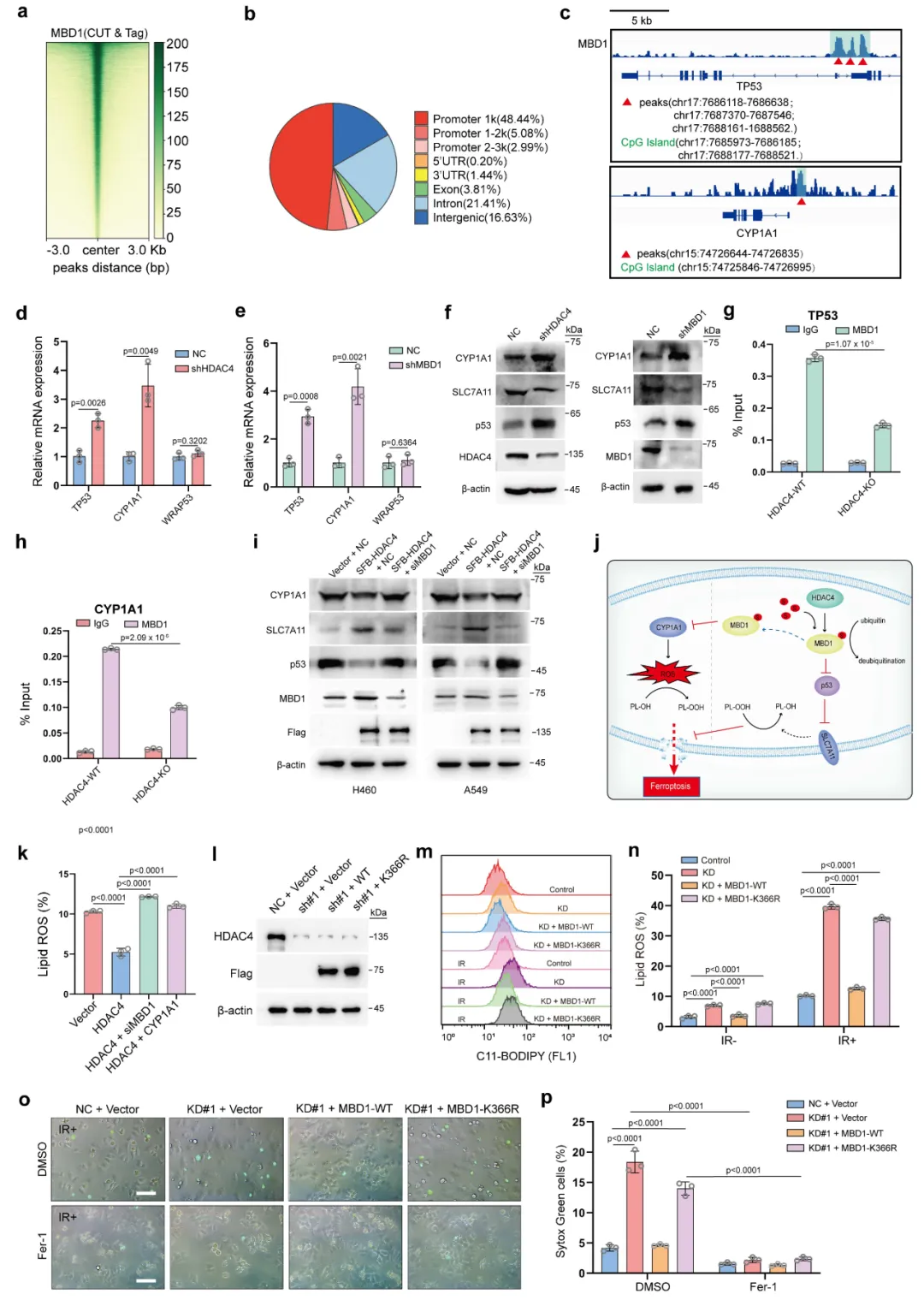
CUT&Tag 实验证明 MBD1 结合并沉默TP53、CYP1A1基因,双重抑制铁死亡信号,由此搭建HDAC4-MBD1-p53/CYP1A1完整耐药调控通路。
本图阐明 MBD1 下游靶基因,串联完整信号通路。首先利用 CUT&Tag 测序(图 5a-b)分析 MBD1 在基因组结合区域,发现其显著富集于TP53、CYP1A1基因启动子区(图 5c)。qPCR 与蛋白检测结果显示,敲低 HDAC4 或 MBD1,TP53、CYP1A1的 mRNA 和蛋白表达均显著上调(图 5d-f);ChIP 实验证明,HDAC4 缺失会减弱 MBD1 在上述基因启动子的结合能力(图 5g-h)。功能回补实验证实,MBD1 可逆转 HDAC4 敲低引发的基因表达变化(图 5i)。细胞功能层面,敲低 HDAC 会提升脂质 ROS,而过表达野生型 MBD1 可恢复表型,K366 突变体则无效(图 5k-p)。
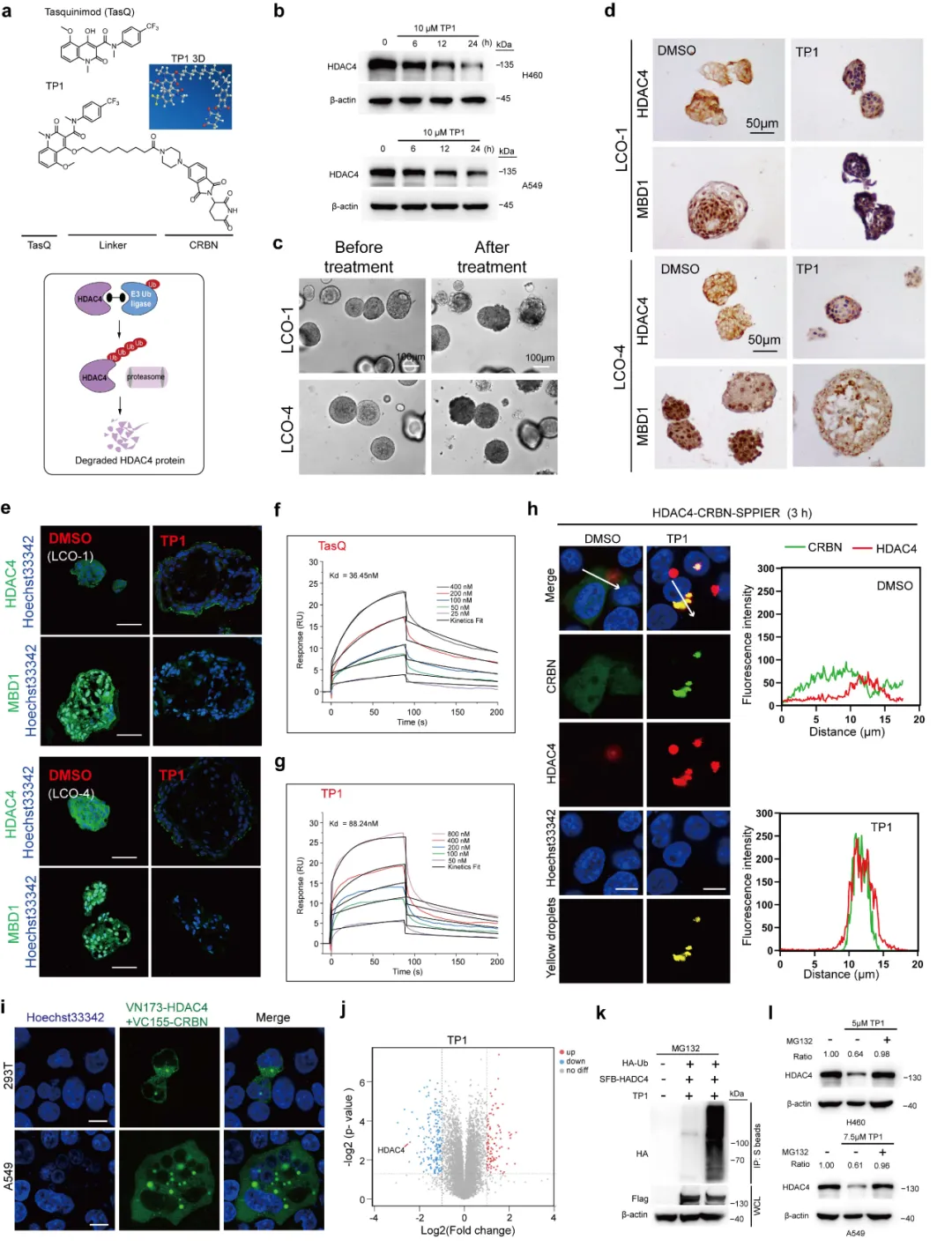
以他喹莫德为母核合成 PROTAC 分子 TP1(图 6),SPR、三元复合物实验证明其可特异性结合并降解 HDAC4。细胞与动物实验(图 7)显示,TP1 下调 HDAC4 及下游蛋白,激活铁死亡,抑瘤与增敏效果全面超越传统抑制剂。
本图围绕基于他喹莫德设计的 HDAC4 特异性 PROTAC 分子 TP1 展开药效与机制研究。图 6a 展示 TP1 化学结构与作用模式:以靶向配体 + 连接子 + CRBN 配体构建 PROTAC,借助泛素 - 蛋白酶体系统降解 HDAC4。时间梯度实验(图 6b)证明 TP1 可时效依赖性降解细胞内 HDAC4,最优作用时长为 24 h。肺癌类器官给药后,HDAC4、MBD1 蛋白同步下调(图 6c-e)。SPR 实验测得 TP1 与 HDAC4 亲和力为 88.24 nM,验证分子结合能力(图 6f-g)。SPPIER、BIFC 成像(图 6h-i)直观观测到 TP1 可诱导HDAC4-CRBN 三元复合物形成,这是 PROTAC 发挥功能的关键。蛋白质组学显示 TP1 降解靶点高度特异(图 6j)。泛素化实验证明 TP1 促进 HDAC4 泛素化(图 6k),加入蛋白酶体抑制剂 MG132 后,降解效应被阻断(图 6l)。
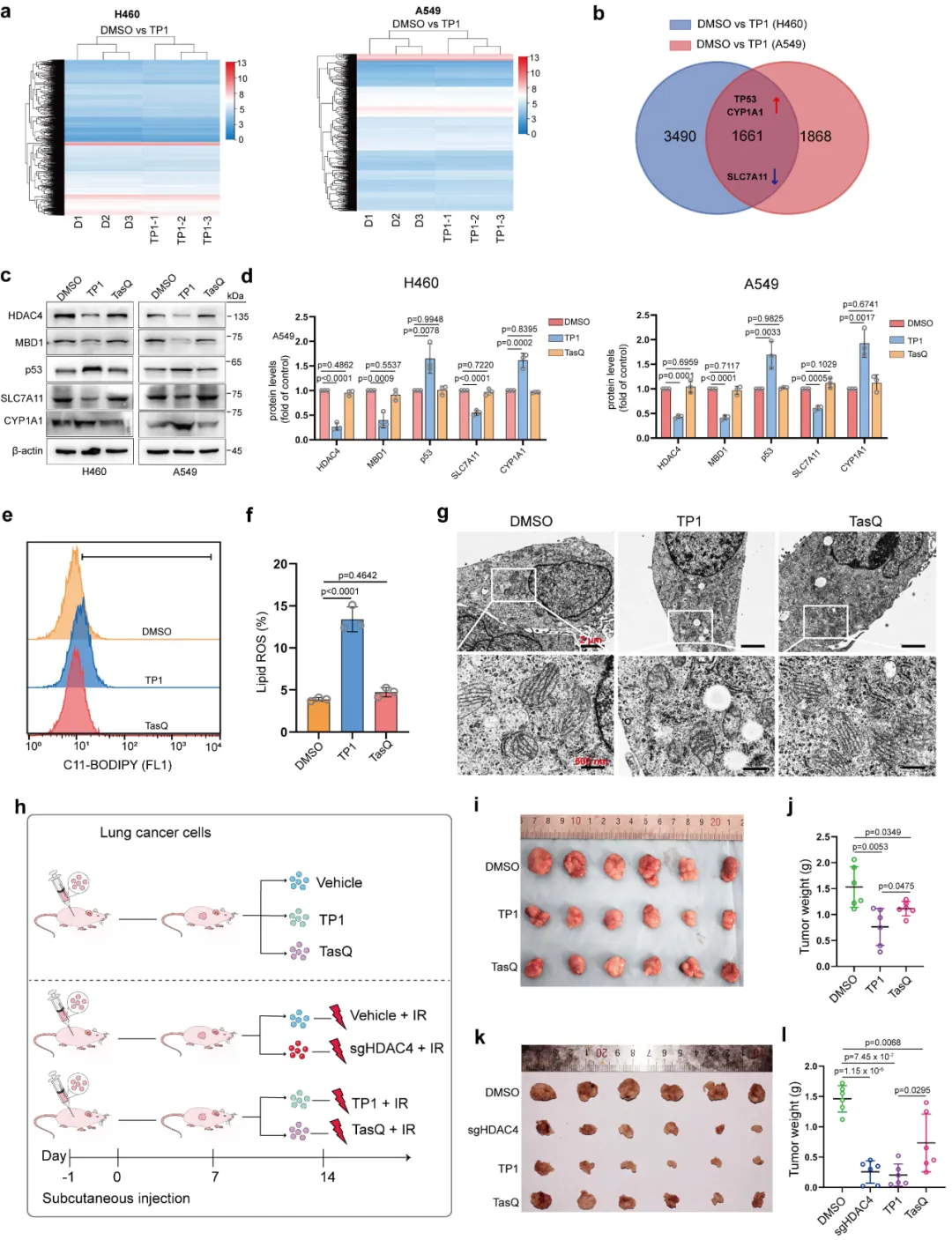
本图从细胞、动物层面对比 TP1 与母体药物他喹莫德(TasQ)的药效差异。转录组分析显示,TP 可上调 p53、CYP1A1,下调 SLC7A11,全面激活铁死亡相关通路(图 7a-b)。蛋白检测(图 7c-d)印证通路分子变化,且 TP1 作用远强于传统抑制剂。C11-BODIPY 检测发现,TP1 处理组脂质 ROS 显著升高(图 7e-f),电镜也观测到典型铁死亡线粒体形态(图 7g)。构建肺癌异种移植瘤模型(图 7h),分组设置溶剂对照、单用 TasQ、单用 TP1、联合放疗等组别。动物给药结果显示,TP1 单药抑瘤效果优于 TasQ;联合放疗后,肿瘤体积、重量被进一步显著抑制(图 7i-l)。同时小鼠体重、肝肾组织病理无异常,证明 TP1 体内安全性良好。
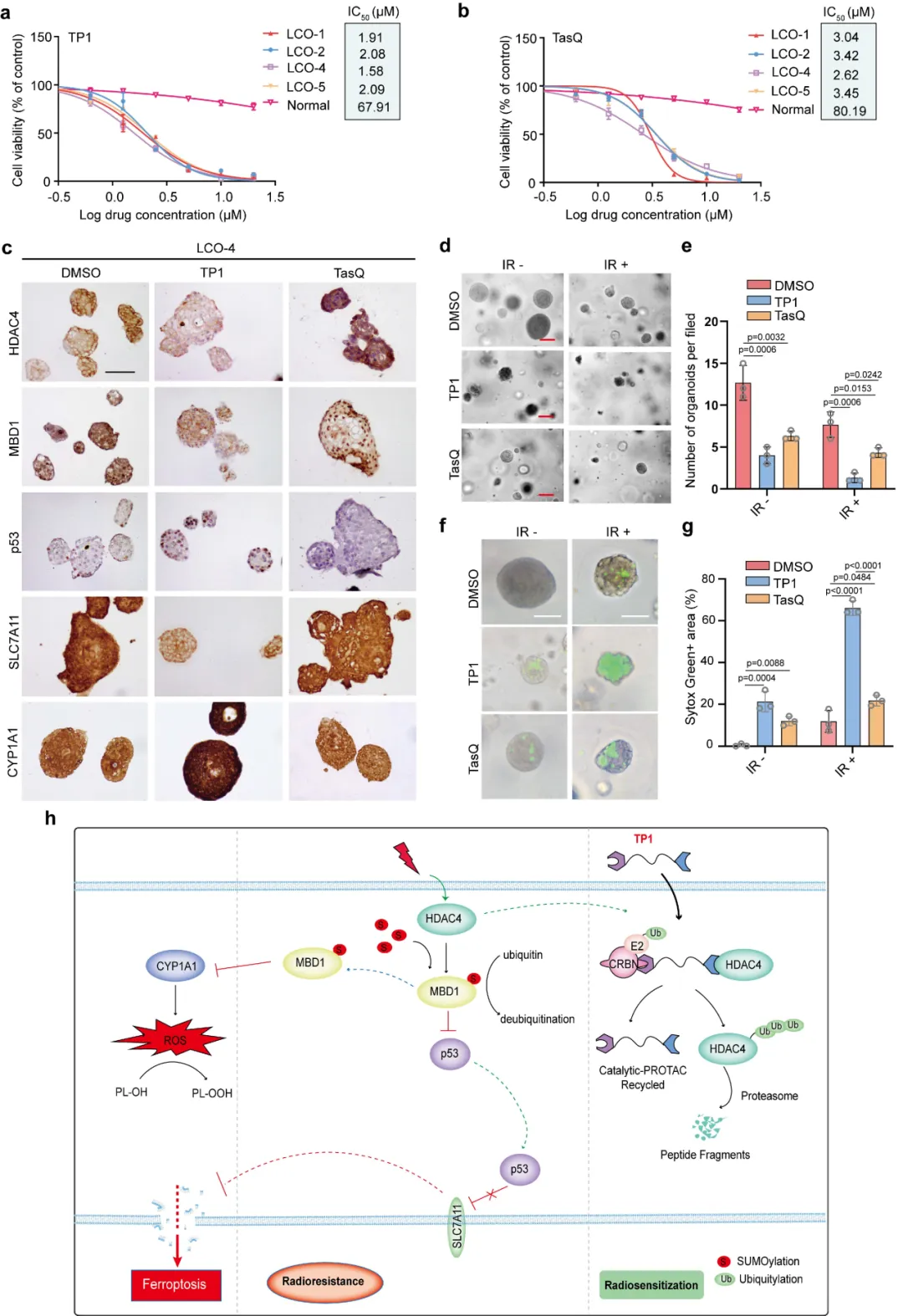
在多株肺癌类器官与正常肺类器官中测评药效,TP1 对肿瘤类器官毒性更强、安全性良好。联合放疗处理后,类器官存活率大幅下降、死亡信号显著增强,直观证明 TP1 可在人体模拟模型中高效逆转放疗耐药,完成从靶点、机制到候选药的全链条验证。
本图以肺癌患者来源类器官(LCO)为核心模型,评估 TP1 的药效、选择性与临床转化价值。图 8a-b 为药物 IC50 曲线:HDAC4 高表达的肺癌类器官对 TP1、TasQ 更敏感,而正常肺类器官药物耐受度高,证明药物具备肿瘤选择性,对正常组织毒性低。免疫组化结果(图 8c)显示,TP1 处理后,类器官内 HDAC4、MBD1、SLC7A11 表达下降,p53、CYP1A1 表达上升,通路变化与细胞、动物实验完全一致。类器官联合放疗实验(图 8d-g):9 Gy 辐射干预下,TP 联合组类器官存活数量最少、SYTOX Green 标记的死亡区域最大,增敏效果显著优于 TasQ 组。最后图 8h 汇总全文调控模型:电离辐射上调 HDAC4,HDAC4 通过 SUMO 化稳定 MBD1,沉默铁死亡相关基因,最终造成放疗抵抗;而 TP1 介导 HDAC4 降解,重启铁死亡,逆转耐药。
本研究明确了 HDAC4 调控肺癌放疗耐药的全新分子机制,开发出靶向 HDAC 的 PROTAC 降解剂 TP1,同时充分证明肺癌类器官在靶点筛选、药效评价中的核心价值。
该成果一方面填补了 HDAC4、蛋白修饰、铁死亡与放疗抵抗之间的机制空白,另一方面为肺癌放疗增敏提供全新候选药物,有望解决临床放疗疗效不佳、肿瘤复发等难题。
后续将继续优化 TP1 分子结构,提升成药性与靶向特异性;开展更多临床样本类器官试验,验证药物普适性;推进免疫健全动物模型试验,探索联合放疗、免疫疗法的协同效果。未来有望推动这款 PROTAC 药物走向临床前研究,为肺癌患者带来新的综合治疗方案。