追求高性能单原子催化剂(SACs)用于氧还原反应(ORR)的研究目前集中于活性位点电子结构调控,而催化剂-电解质界面及相应水结构的关键影响却未得到足够关注。
2026年04月24日,南昌大学陈义旺、袁凯团队在Energy & Environmental Science期刊发表题为“Beyond electronic effects: hydrogen-bond engineering via N-mediated microenvironment control for accelerated oxygen electroreduction”的研究论文,团队成员姜文惠为论文第一作者,陈义旺、袁凯为论文共同通讯作者。
第一作者:姜文惠
通讯作者:陈义旺、袁凯
通讯单位:南昌大学
论文DOI:10.1039/D6EE00569A
该研究开发了基于钴卟啉聚合物的分子精确平台,其连接基团上具有原子精确的N梯度分布。这种无热解策略通过连接基团上的N原子同时调控了Co-N₄位点的电子结构,同时利用N原子的强电负性来锚定和重构界面水分子。通过整合的原位光谱分析、从头算分子动力学AIMD模拟以及大量实验证据,证明了富N连接基团优化了界面氢键网络,促进了质子通过Grotthuss机制的高效传输,并显著降低了速率决定性质子耦合电子转移步骤(*OOH形成)的能垒。因此,吡嗪连接催化剂(PZ-POP-Co)在ORR中的半波电位显著提高了80 mV,并且其在锌空电池中的峰值功率密度是其无N对应物的3倍。该研究确定了界面氢键网络工程是高性能SACs一项关键但迄今被忽略的准则,为推进质子耦合电催化提供了新视角。
氧还原反应(ORR)是包括燃料电池和金属空气电池在内的电化学能源转换器件的基础,但其多步质子耦合电子转移(PCET)过程导致反应动力学缓慢和过电位高,从而限制了器件的输出功率和能量转换效率。开发活性电催化剂以确保高效的ORR动力学是应对这一挑战的关键步骤。尽管铂基催化剂表现出卓越的催化活性,但其固有的稀缺性和耐久性限制使得人们需要高性能的非贵金属替代品。
具有M-Nₓ配位结构的非贵金属基单原子催化剂(SACs),以其最大化的原子利用率和成本效益为特征,已成为有前景的解决方案。虽然大量研究集中于优化其电子结构,但催化剂-电解质界面及其相关氢键网络的关键作用却被忽视。对于水性电解质中的ORR,催化剂-电解质界面处的质子转移主要通过Grotthuss机制进行,其中质子在由H₂O主导的动态互联氢键网络中跳跃并转移到电极表面。界面氢键网络主要控制着界面处的质子转移动力学。然而,传统碳化SACs的结构复杂性掩盖了清晰的结构-性能关系,留下了分子水平理解上的关键空白。因此,解析和控制界面水结构不仅是电催化ORR的基础,也是开发具有优异活性和稳定性的先进催化剂的必要条件。
在此,该研究通过分子定义的卟啉平台弥合了这一长期存在的空白,该平台避免了热解引起的不确定性。受金属酶(如细胞色素c氧化酶)的启发,基于钴卟啉的共价有机聚合物为通过轨道杂化对Co-N₄中心进行电子调控,同时通过分子水平的界面水工程实现氢键网络重构,提供了一个理想的模型系统。利用无热解策略,合成了在连接位点具有原子精确N梯度分布的钴卟啉基聚合物框架(X-POP-Co;X = 苯(BDA)、吡啶(PD)、吡嗪(PZ))。该方法保留了Co-N₄配位构型,同时系统调控了电子密度分布,并利用N的强电负性通过定向氢键调控界面水结构。原位衰减全反射表面增强红外光谱(ATR-SEIRAS)结合从头算分子动力学AIMD模拟和密度泛函理论DFT计算,决定性地证明了富N连接基团通过优化亲水区域增加了界面H₂O浓度,并形成了稳健的氢键网络以促进质子通过Grotthuss机制的跳跃,从而降低了限速PCET能垒。因此,吡嗪连接催化剂(PZ-POP-Co)实现了卓越的ORR活性,其半波电位比其无N连接对应物提高了80 mV,而组装的锌空电池(ZAB)的峰值功率密度提高了约3倍,循环稳定性延长了2倍。该研究提供了机理性的论证,将界面氢键网络工程确立为SACs设计中缺失的关键环节,超越了传统的电子结构调控范式,并从界面氢键网络的角度为优化质子耦合电催化过程提供了新策略。
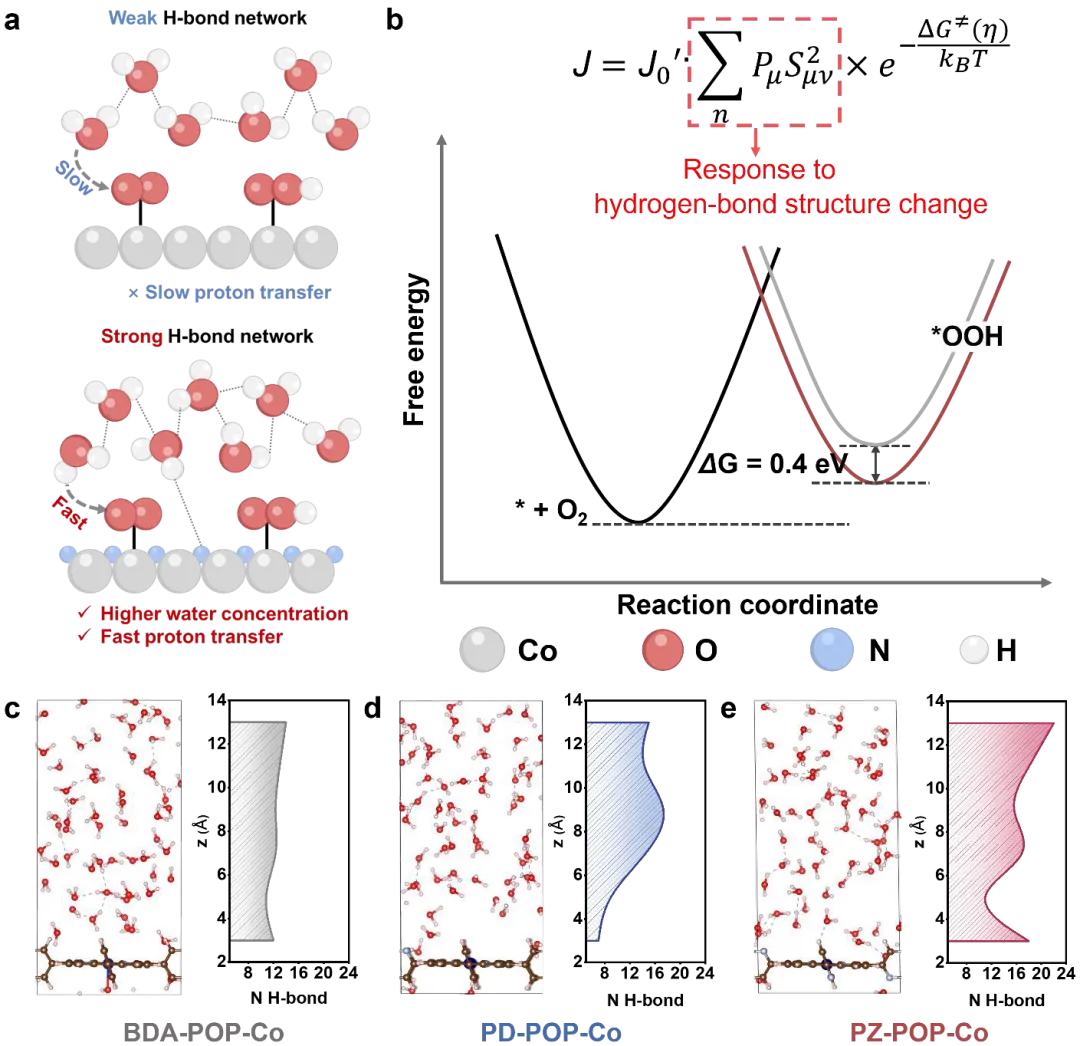
图1. 氮原子对界面氢键网络的调控。(a) 氮介导的氢键网络促进界面水捕获和质子转移的示意图。(b) X-POP-Co的设计概念。Pμ是与反应物态μ对应的玻尔兹曼布居数,S²μν代表质子平方的振动耦合,ΔG≠是克服活化自由能的玻尔兹曼概率。电流密度表达式表明,J₀(交换电流密度)正比于PμS²μν,而该值由氢键结构主导。kB,玻尔兹曼常数。通过AIMD模拟得到的(c)BDA-POP-Co、(d)PD-POP-Co和(e)PZ-POP-Co的界面水分子取向。
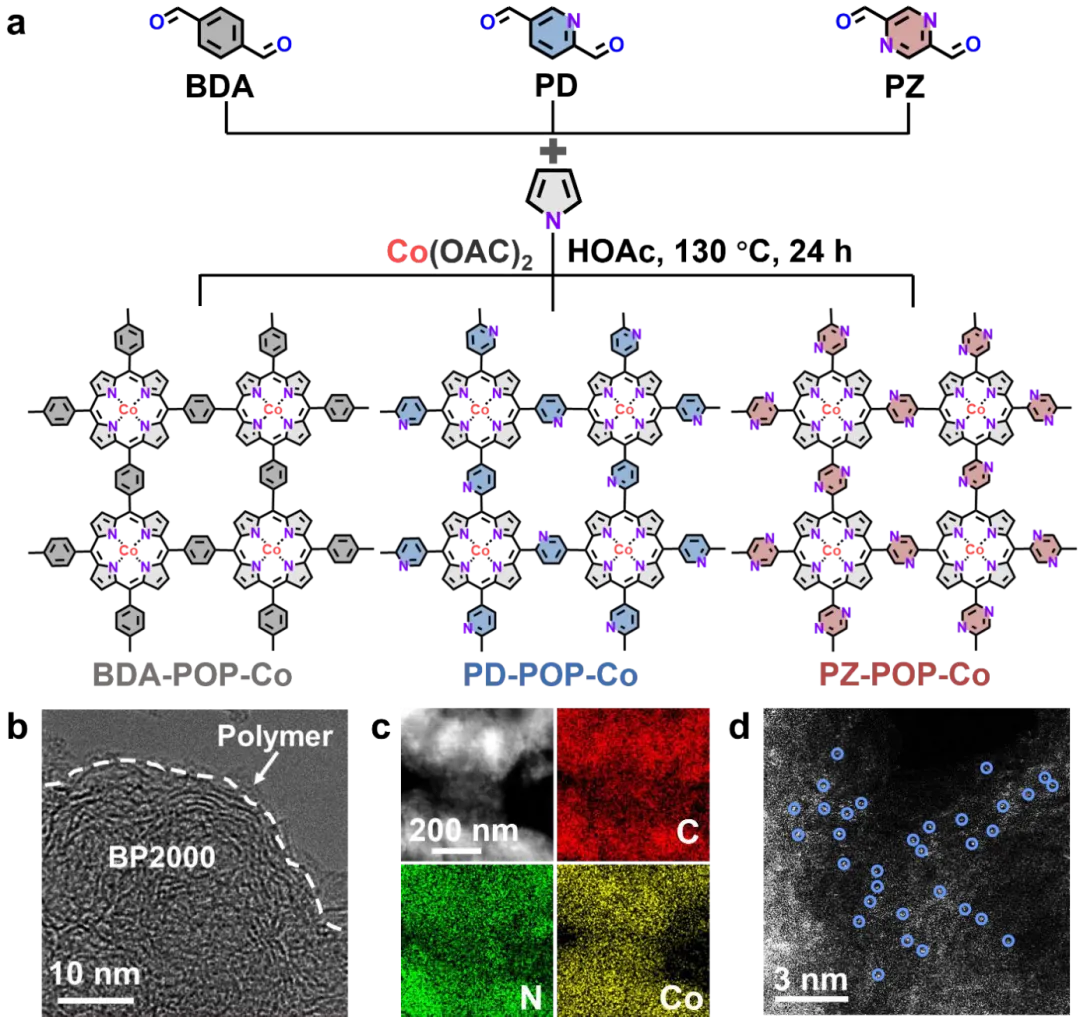
图2. X-POP-Co的设计原理与结构表征。(a) X-POP-Co合成的示意图。(b) PZ-POP-Co的高分辨率TEM图像,以及(c)带有相应EDS元素面分布图的TEM图像。(d) PZ-POP-Co的像差校正HADDF-STEM图像(孤立的Co原子用蓝圈标出)。
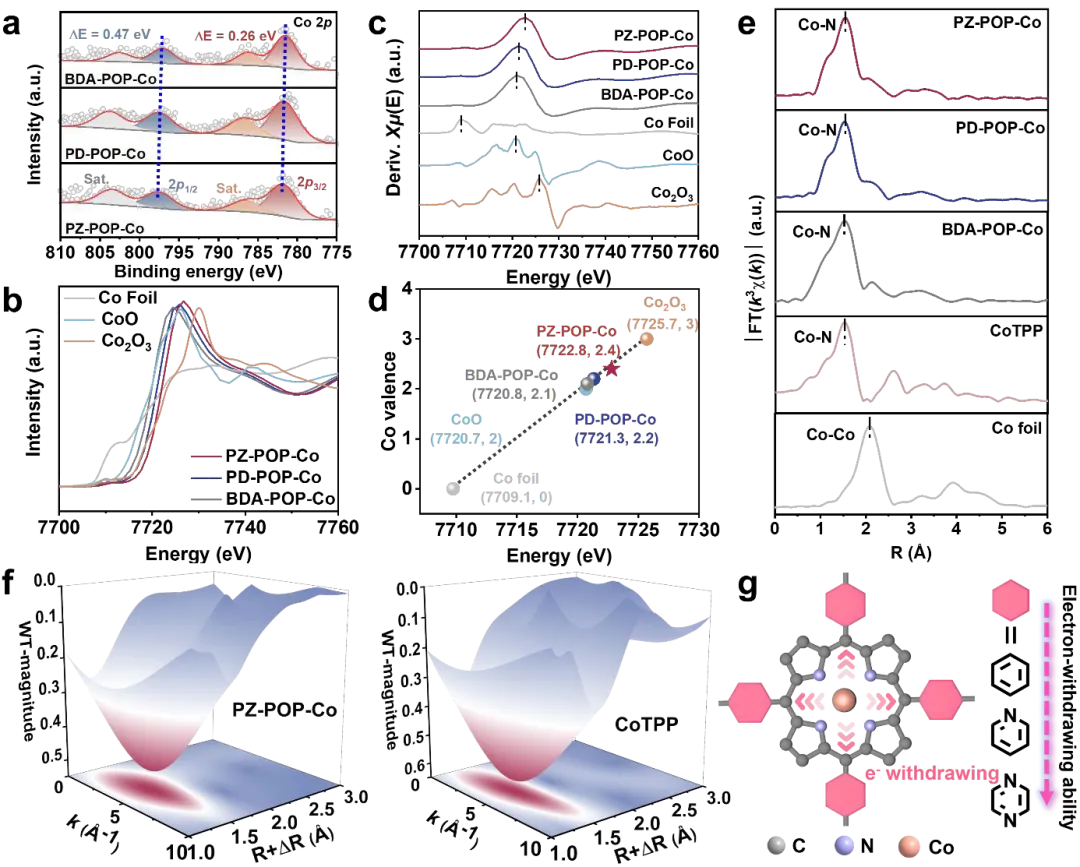
图3. X-POP-Co类似物中Co中心的电子构型与局部配位。(a) X-POP-Co类似物的高分辨率Co 2p XPS谱。(b) 归一化的Co K边XANES谱和(c)它们对应的一阶导数谱。(d) 通过相对于Co箔、CoO和CO₂O₃参比的线性拟合校准,定量测定X-POP-Co中Co的平均氧化态。(e) X-POP-Co、Co箔、CoO和CoTPP的k3加权Co K边EXAFS谱的傅里叶变换幅度谱。(f) PZ-POP-Co和CoTPP的WT-EXAFS图。(g) 不同连接基团吸电子效应的示意图。
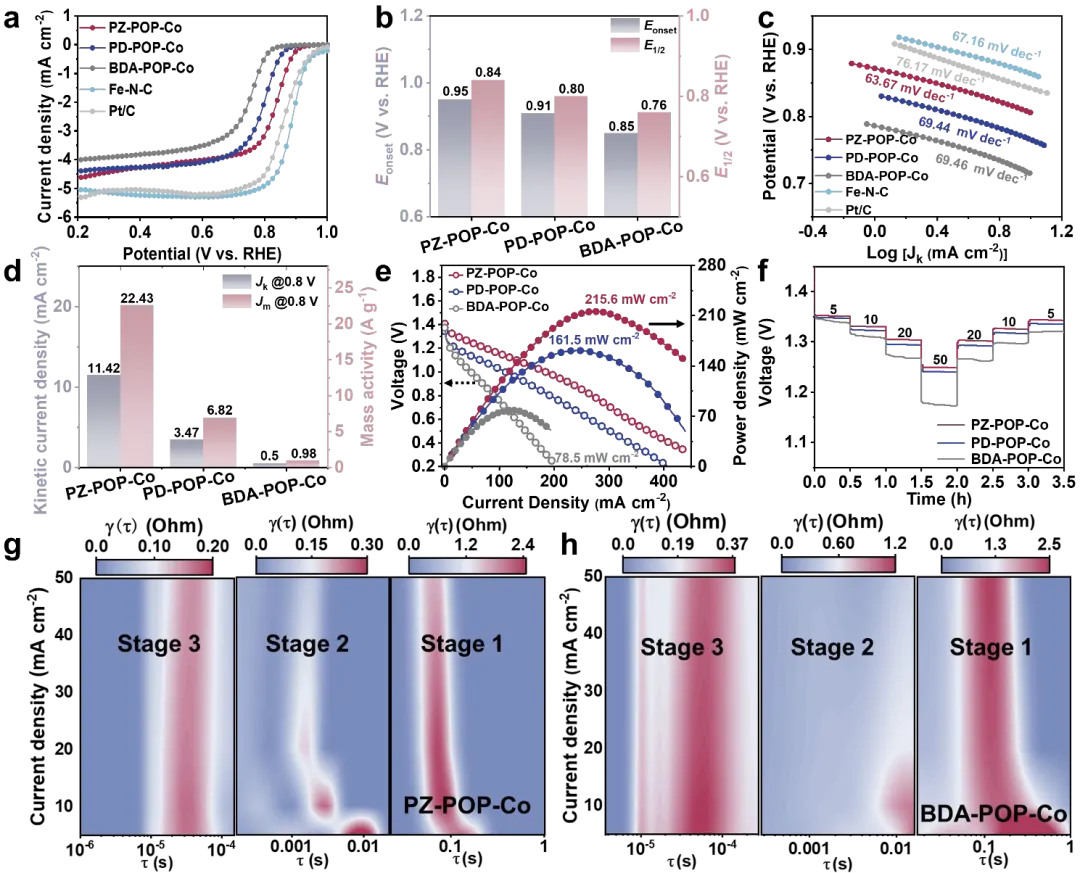
图4. ORR性能评估。(a) X-POP-Co、Fe-N-C和Pt/C催化剂在O₂饱和的0.1 M KOH溶液中、1600 rpm转速下的ORR极化曲线。(b) X-POP-Co催化剂的Eonset和E₁/₂。(c) X-POP-Co、Fe-N-C和Pt/C催化剂的塔菲尔曲线。(d) X-POP-Co催化剂在0.8 V下的Jₖ和Jₘ。(e) 基于X-POP-Co的ZAB的放电极化曲线及相应的功率密度图。(f) 不同电流密度下的恒流放电曲线。(g) 基于PZ-POP-Co和(h)BDA-POP-Co催化剂的液态ZAB的DRT结果等高线图。
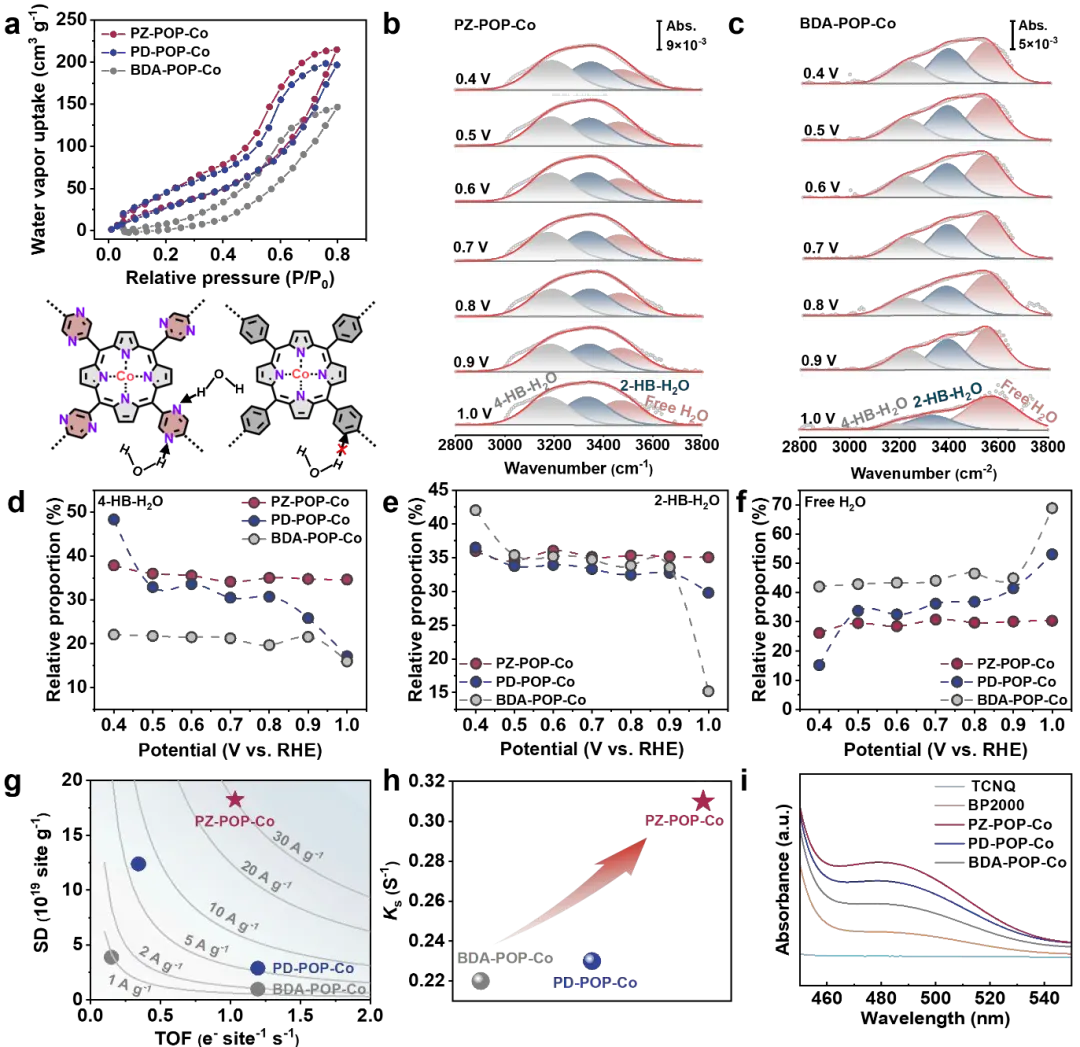
图5. 质子与电子转移机制研究。(a) X-POP-Co的水吸附等温线。(b) PZ-POP-Co和(c)BDA-POP-Co在不同电位下界面水的O-H伸缩振动的原位ATR-SEIRAS谱。(d-f) 不同类型界面水的相对比例。(g) 基于0.8 V下SD和TOF值的X-POP-Co的ORR等活性图。(h) X-POP-Co的界面电子转移动力学常数。(i) 在BP2000和X-POP-Co存在下,乙腈中TCNQ溶液的UV-vis光谱。
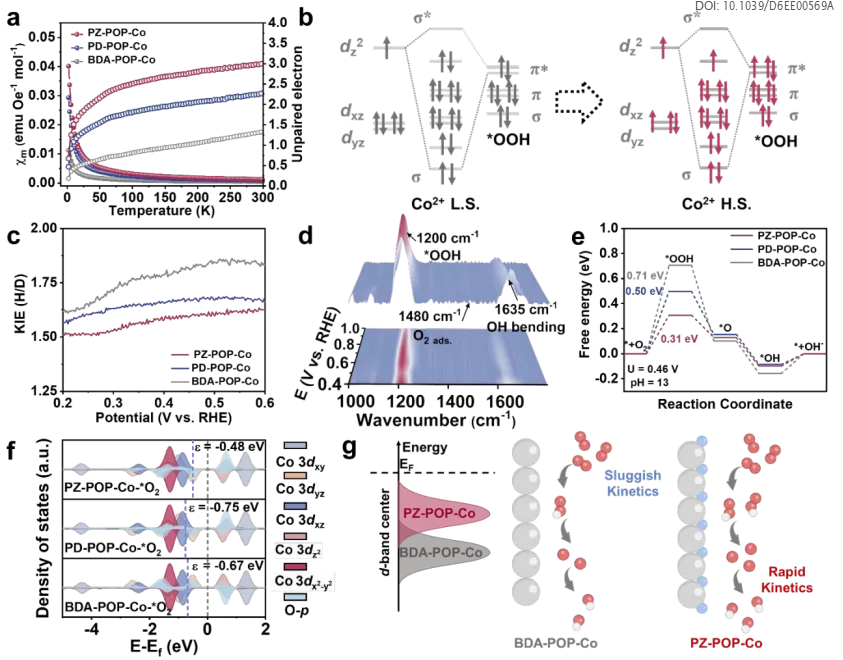
图6. ORR机理分析。(a) X-POP-Co的磁化率。(b) 不同自旋态下*OOH与Co中心之间不同轨道相互作用的示意图。(c) X-POP-Co的KIE。(d) PZ-POP-Co在1000-1800 cm⁻¹范围内ORR的原位ATR-SEIRAS谱。(e) U = 0.46 V时X-POP-Co类似物的吉布斯自由能图。(f) 吸附在X-POP-Co类似物上的O₂的投影态密度。(g) BDA-POP-Co和PZ-POP-Co吸附O₂后的d带中心及整体ORR动力学示意图。
总之,该研究通过一个无热解、分子级定义的卟啉框架平台,揭示了氮掺杂剂在碳基ORR催化剂中的关键作用,提供了超越传统认知的机理见解。该研究证明了富氮连接基团同时优化了Co-N₄位点的电子结构并重构了界面氢键网络,产生亲水区域以提高局部水浓度并促进高效质子转移。由此产生的动态平衡氢键结构将RDS(*OH中间体的形成)能垒降低了超过56%,决定性地加速了PCET动力学。因此,吡嗪连接催化剂(PZ-POP-Co)在碱性条件下比其无氮对应物高出80mV的半波电位。当集成到锌-空气电池中时,PZ-POP-Co的峰值功率密度提高了3倍(215.6mW cm⁻²),循环稳定性延长了2倍。该研究提供了机理见解,将界面氢键网络工程确立为高性能单原子催化剂的一个关键但此前被忽视的设计原则,为推进质子耦合电催化提供了通用蓝图。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
