赣州市妇幼保健院确诊一例极罕见脂肪酸氧化代谢病——肉碱棕榈酰转移酶1缺乏症

近日,赣州市新生儿疾病筛查中心(赣州市妇幼保健院)首次通过串联质谱技术筛查确诊一例先天性遗传代谢病——肉碱棕榈酰转移酶1缺乏症(Carnitine Palmitoyltransferase I Deficiency),此病例极为罕见。
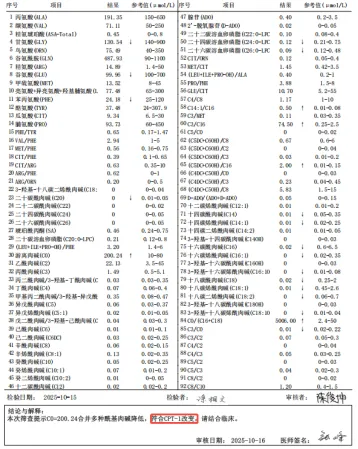
该患儿出生后无特殊临床表现,出生满48小时后,在分娩机构采集足底血,送赣州市新筛中心(赣州市妇幼保健院)接受新生儿疾病筛查。串联质谱多种遗传代谢病筛查结果显示C0升高(200.24μmol/L)合并多种酰基肉碱降低,符合CPT-1改变,提示可能存在脂肪酸代谢障碍。新筛中心立即启动召回流程,通过召回复查,遗传代谢门诊结果仍提示异常。经进一步基因检测分析,最终检出患儿CPT1A基因的致病变异,确诊为肉碱棕榈酰转移酶1缺乏症。
肉碱棕榈酰转移酶1缺乏症是一种发病率极低的常染色体隐性遗传病,由于CPT1A基因突变,导致肉碱棕榈酰转移酶I功能缺陷。据流行病学统计,德国西南地区1084195名新生儿中发现1例,葡萄牙316243名新生儿中发现1例,日本200000名新生儿筛查出1例。
该酶是长链脂肪酸进入线粒体进行能量代谢的关键酶。肉碱棕榈酰转移酶1缺乏症临床表现,患者首次出现症状大多集中在出生后数小时至30个月。饥饿和感染性疾病是常见诱因,发病急,死亡率高。典型表现有低酮型低血糖或肝性脑病所致的呕吐、意识改变、惊厥、昏迷、肝大伴转氨酶升高、凝血功能异常,以及血氨、血脂增高等。可伴有酸中毒、碱性尿、磷酸盐排出增多,提示肾小管性酸中毒,脑部远期损害主要取决于低血糖的严重程度。
该病例确诊得益于家长在常规免费筛查项目基础上,选择增加串联质谱多种遗传代谢病筛查。与传统筛查方法不同,串联质谱技术可高效检测此类脂肪酸氧化障碍疾病。虽然该病迄今无法根治,但早期诊断并配合科学饮食管理(如避免长时间饥饿、低脂高糖饮食、规律进食)及必要时的中链甘油三酯补充,可有效预防危象发生,保障患儿正常生长发育。若未能及时诊断,首次严重代谢危象发作可能对神经系统造成不可逆损伤。
①先天性甲状腺功能减低症(CH)
②苯丙酮尿症(PKU)
③先天性肾上腺皮质增生症(CAH)
④葡萄糖-6-磷酸脱氢酶缺乏症(G6PD)
①串联质谱多种遗传代谢病筛查(可检测近50种遗传代谢病)
②地中海贫血筛查
③脊髓性肌萎缩症(SMA)基因筛查
绝大多数遗传代谢病早期无明显症状,但隐藏的风险可能在特定诱因下突然暴露。串联质谱技术实现“一滴血筛查多种疾病”,为早期干预赢得宝贵时间。本例患儿的成功确诊充分体现了扩展性新生儿筛查对儿童健康的重要意义。
三审(校):李跃琼为我们点亮『星标』