南昌大学第二附属医院心肌纤维化新调控轴:MYC-METTL1 介导 m7G 修饰调控 HILPDA 驱动心梗后心室重构
- 2026-06-28 16:35:18
文献学习
本研究针对心梗后心肌纤维化表观转录调控机制尚不明确的科学问题,先利用公共数据库生物信息学预测 MYC、METTL1、HILPDA 三者存在表达关联与结合位点,再构建小鼠心梗在体模型与缺氧 / TGF-β1 诱导心脏成纤维细胞离体模型,依次运用分子互作、基因过表达 / 敲低、组织病理、心功能检测等多组实验验证上下游调控逻辑:先证实心梗后 HILPDA 特异性在成纤维细胞上调并促线粒体损伤与成纤维细胞活化,再阐明 METTL1 通过 m7G 修饰稳定 HILPDA mRNA 提升其蛋白水平,最后证实转录因子 MYC 直接结合 METTL1 启动子激活其转录,体内纤维化细胞特异性敲低 HILPDA 可逆转线粒体功能障碍、减少胶原沉积、缩小梗死面积并恢复心功能,完整证实 MYC-METTL1-HILPDA 轴是心梗后心肌纤维化全新驱动通路,为心衰防治提供表观转录与代谢相关新型治疗靶点。
研究背景
心肌梗死会诱发心肌纤维化,持续纤维化造成心室重构与心力衰竭,既往研究多聚焦 TGF-β 等经典通路,心梗后纤维化的表观转录调控机制仍缺乏系统阐释;m7G 甲基化修饰可调控 RNA 稳定性,METTL1 作为核心 m7G 甲基转移酶被报道参与心肌缺血损伤,但下游靶基因未知;脂质代谢调控蛋白 HILPDA 可诱导线粒体功能紊乱,在肝肾纤维化中发挥促纤维化作用,其在心肌纤维化中的功能及上游调控通路未被解析;转录因子 MYC 在多器官纤维化中升高,生物信息提示 MYC 可能调控 METTL1 转录,三者形成的调控轴在心梗纤维化中的作用完全未见报道,因此本研究围绕该调控轴展开机制探究。
研究方案
首先依托公共数据库开展生物信息分析,预测 HILPDA 存在 m7G 修饰位点、METTL1 与 HILPDA 存在结合、MYC 可结合 METTL1 启动子且三者表达正相关,提出 MYC-METTL1-HILPDA 调控轴假说;其次构建小鼠心梗在体模型与缺氧、TGF-β1 诱导心脏成纤维细胞离体模型,通过时序表达实验锁定 HILPDA 表达峰值;再通过基因过表达、siRNA/AAV 敲低分别验证 HILPDA、METTL1、MYC 的细胞功能;借助 RIP、ChIP、双荧光素酶、放线菌素 D 降解实验验证分子间结合与 mRNA 稳定机制;结合组织染色、心功能检测、线粒体功能、细胞增殖迁移实验逐级解析上下游调控逻辑;最后体内特异性敲低 HILPDA 验证通路靶向干预的心脏保护效果。
研究结果
心梗小鼠心肌组织与原代心脏成纤维细胞中 METTL1、HILPDA、MYC 表达显著上调,HILPDA 于心梗后 7 天达到表达峰值;HILPDA 主要在心脏成纤维细胞发挥作用,过表达会诱导线粒体 ROS 升高、膜电位下降、ATP 合成减少,促进成纤维细胞增殖迁移与肌成纤维细胞转化,敲低则逆转上述损伤;METTL1 通过 m7G 修饰结合并稳定 HILPDA mRNA,提升 HILPDA 蛋白表达,缺失 HILPDA 可抵消 METTL1 促纤维化效应;MYC 直接结合 METTL1 启动子促进其转录,MYC 上调同步升高 METTL1、HILPDA 与整体 m7G 修饰水平;成纤维细胞特异性 AAV 敲低 HILPDA 可改善心梗小鼠左室射血分数、缩小梗死面积、减轻胶原沉积、恢复线粒体功能,且不反馈调控上游 MYC、METTL1。
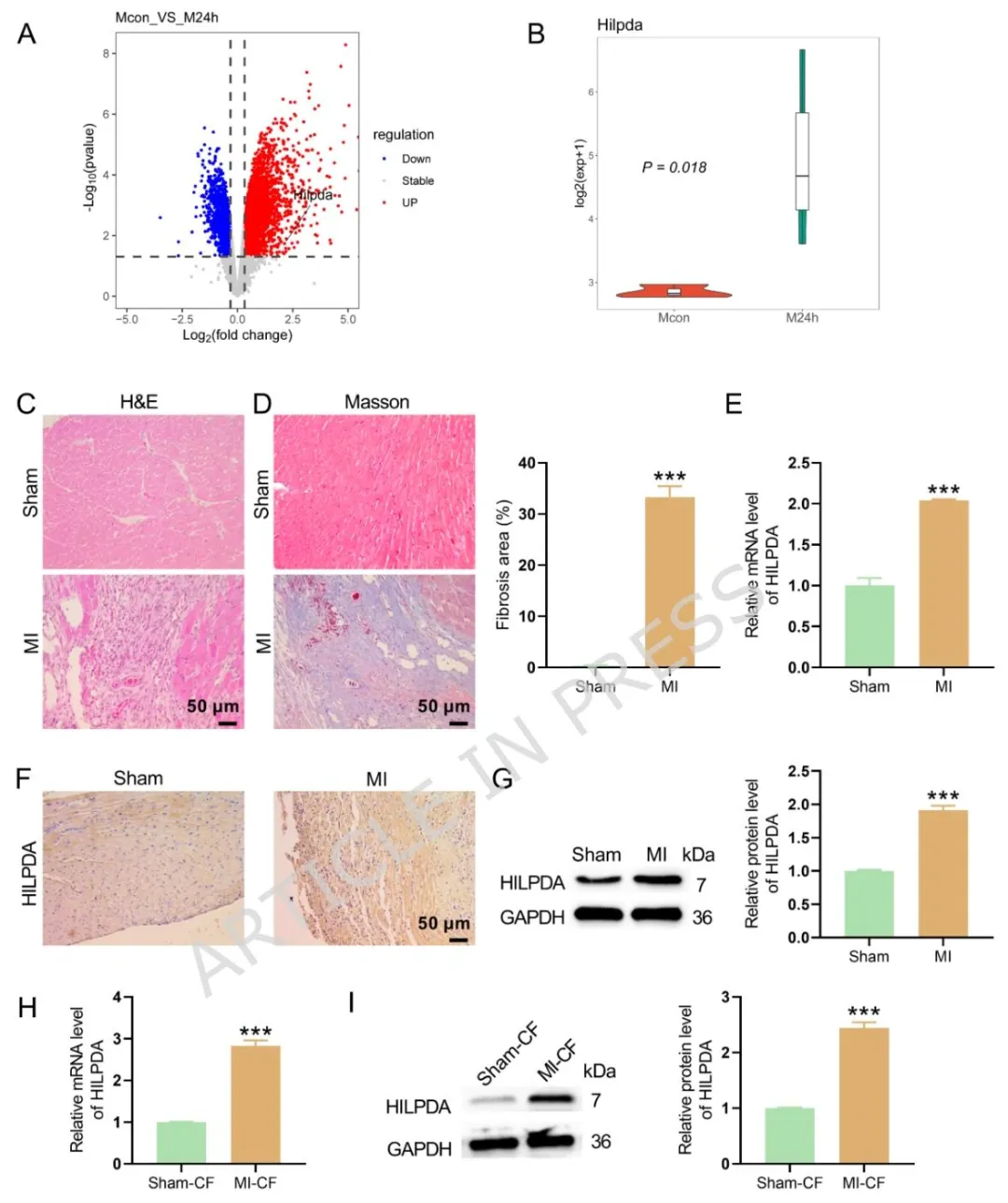
Figure1:公共数据集与小鼠心梗时序模型验证心梗后心肌组织及原代成纤维细胞内 HILPDA 显著上调,同时病理染色证实心梗组存在心肌坏死、炎症浸润与胶原沉积,初步提示 HILPDA 参与心梗纤维化进程。
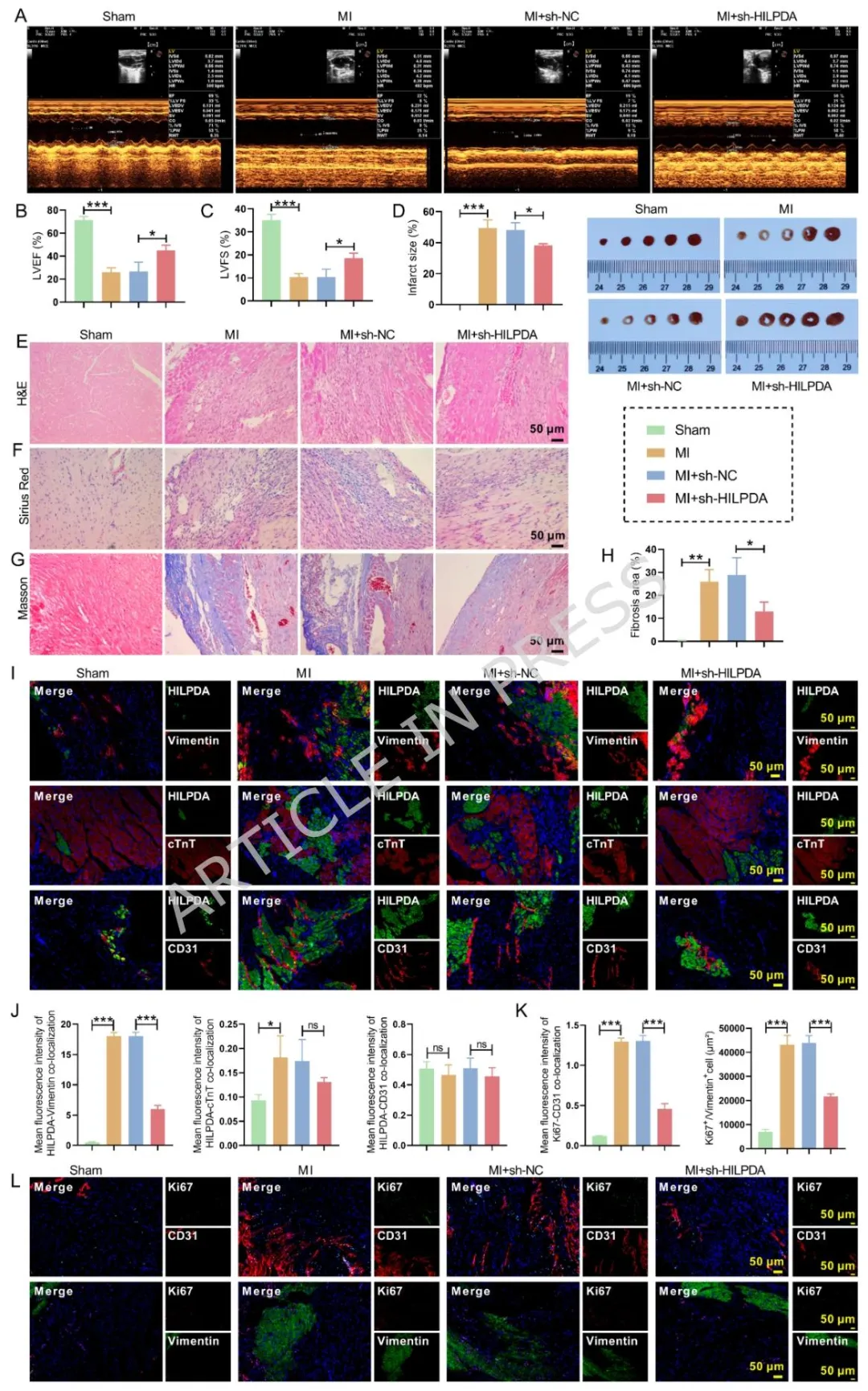
Figure2:体内成纤维细胞特异性敲低 HILPDA 可改善心梗小鼠心功能、缩小梗死面积,减轻心肌病理损伤与胶原沉积,共定位染色证明 HILPDA 主要表达于成纤维细胞,敲低后显著抑制成纤维细胞与内皮细胞增殖,明确 HILPDA 体内促纤维化作用。
Figure3:体外缺氧诱导成纤维细胞模型证实 HILPDA 过表达加剧线粒体功能损伤、脂质蓄积、细胞增殖迁移及肌成纤维细胞活化,敲低可逆转全部缺氧诱导的促纤维化表型,明确 HILPDA 介导线粒体紊乱驱动成纤维细胞活化。
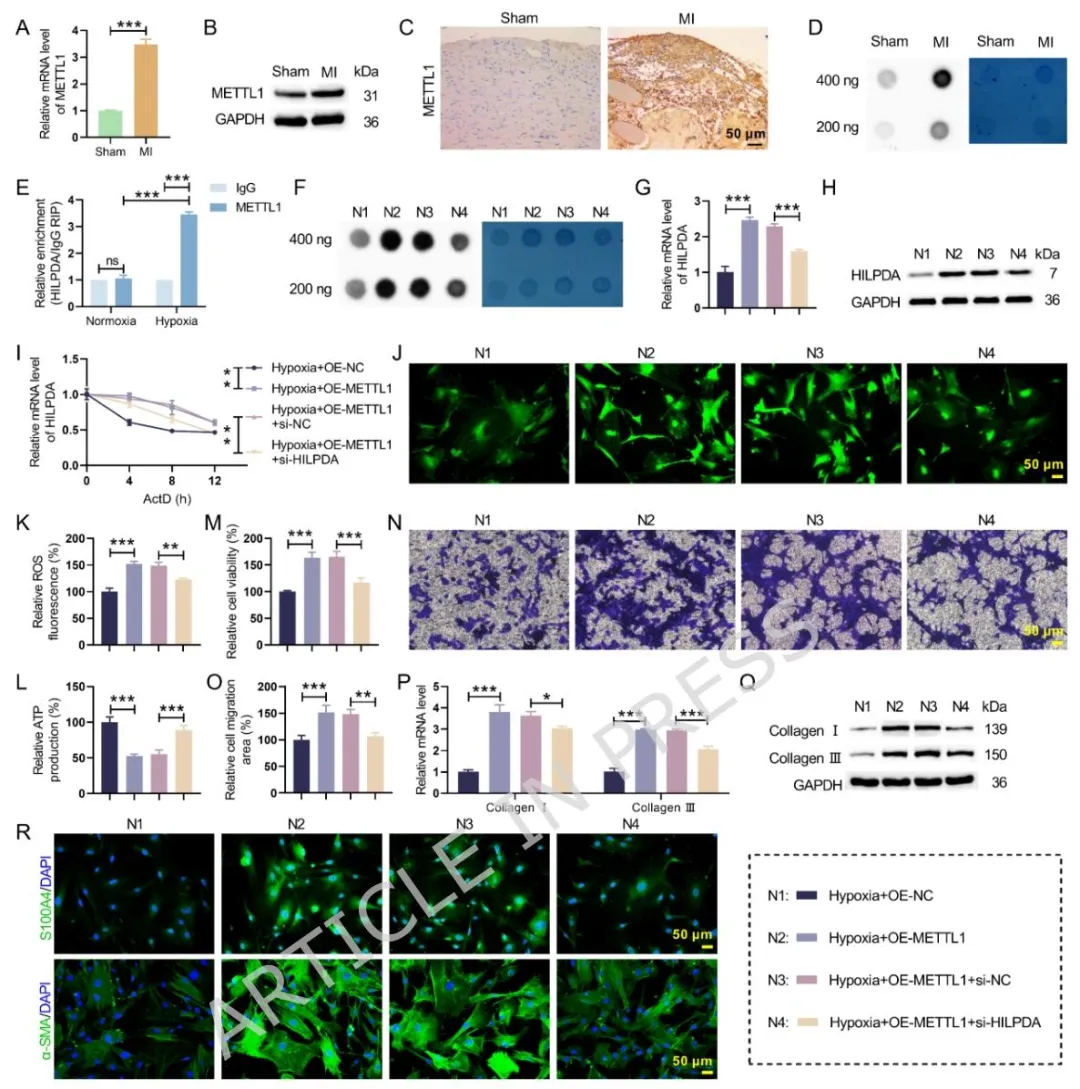
Figure4:体内验证心梗后 METTL1 与整体 m7G 修饰水平升高,体外 RIP 证实 METTL1 结合 HILPDA mRNA;METTL1 过表达通过 m7G 修饰提升 HILPDA mRNA 稳定性,加重线粒体损伤与纤维化表型,敲低 HILPDA 可完全阻断 METTL1 的促纤维化效应,阐明 METTL1 的下游效应分子为 HILPDA。
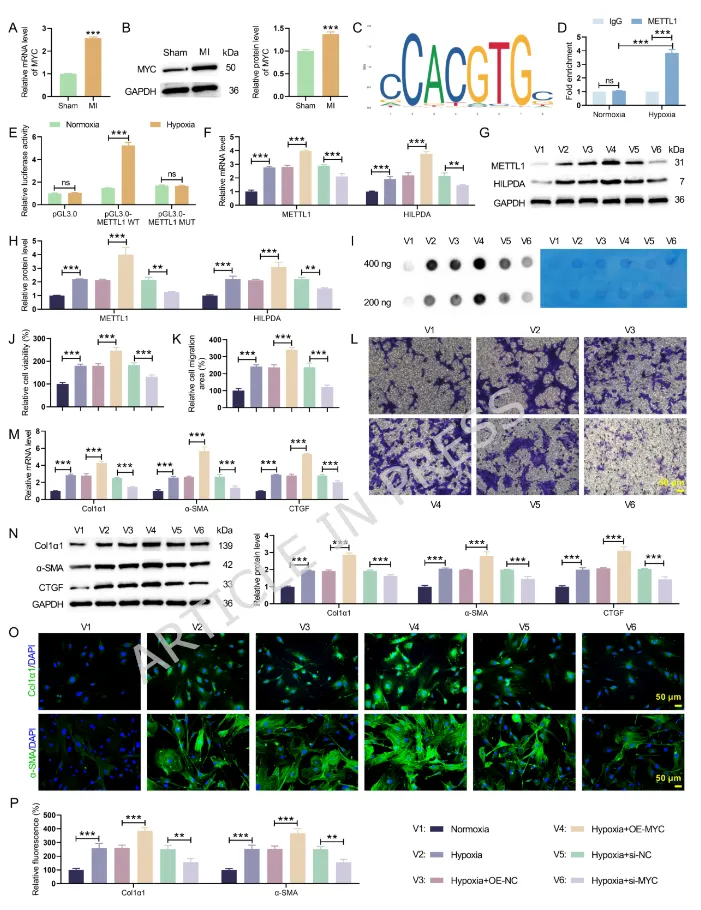
Figure5:生物信息、ChIP、双荧光素酶实验证实 MYC 直接转录激活 METTL1;MYC 过表达同步上调 METTL1、HILPDA 与 m7G 修饰水平,加剧成纤维细胞活化,敲低 MYC 抑制整条通路活化,确立 MYC 为 METTL1 上游转录调控因子。
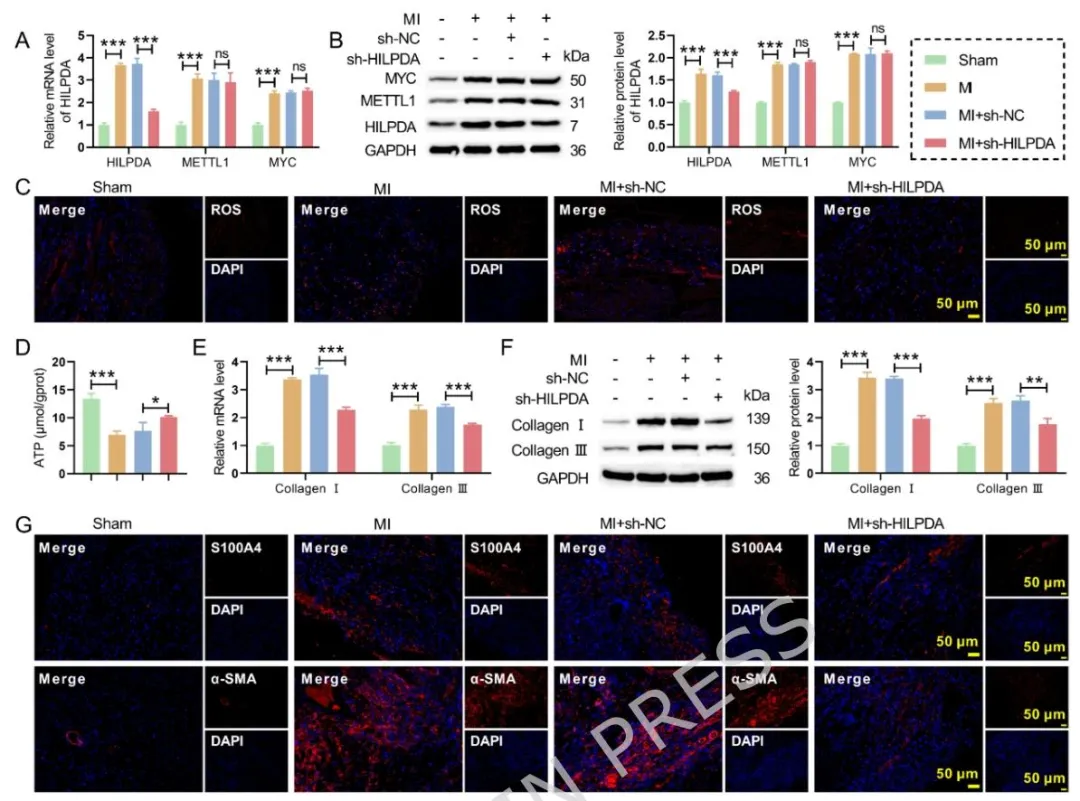
Figure6:体内敲低 HILPDA 不改变 MYC、METTL1 表达,仅降低 HILPDA 水平,同时缓解心梗后心肌 ROS 蓄积、ATP 合成不足,下调胶原与成纤维细胞活化标志物,反向验证 HILPDA 是该通路发挥心脏损伤效应的关键执行分子。
研究结论
本研究完整阐释 MYC-METTL1-HILPDA 轴作为心梗后心肌纤维化全新驱动通路的分子机制,串联转录调控、m7G 表观转录修饰、脂质代谢与线粒体功能紊乱多层次调控网络,解释了心梗后持续性纤维化的分子诱因,补充了心血管领域 m7G 修饰的纤维化相关研究,同时证实靶向 HILPDA 具备心脏保护与抗纤维化治疗潜力,为心衰干预提供新靶点;研究也存在明显局限,尚未精准定位 METTL1 介导的 HILPDA mRNA 具体 m7G 修饰位点及其功能差异,未阐明 HILPDA 通过脂质代谢诱导线粒体损伤的下游分子通路,也未探究该通路在其他心肌病模型中的普适性;后续可通过质谱与基因编辑定位修饰位点,借助脂质组学解析 HILPDA 线粒体调控机制,拓展多疾病模型验证通路作用,并开发通路小分子抑制剂推动临床转化研究。
本文中使用的图片来源Pubmed,因客观原因未能与权利人取得联系。本平台出于学术交流目的引用,无意侵犯原作者权益。如权利人认为不妥,请及时联系公众号后台,我们将立即删除或协商解决。
国家杰青一对一答疑视频
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 南昌市“洪城红”青年应急队江西服装学院中队以实际行动彰显青春担当!
- 南昌茅台老酒高价回收,实在经营不坑不骗,随叫随到
- 南昌汽车救援搭电一键呼叫19979009448【东湖区西湖区青云谱区红谷滩区】车主速存!24 小时全城道路救援,汽车抛锚不用慌
- 坐标南昌,明装暖气片完美落地!
- 南昌赣江边那棵探水老樟被游客拍爆,本地人路过冷笑:这树前几年就烂芯了,不怕哪天顺着江坡翻下去?
- 南昌人要享福了!青山湖即将开启快速通道时代,生活在这儿的人都要乐不可言了!
- 【南昌招聘】南昌县总医院公立医院总量管理人员招聘公告
- 独家曝出!南昌青山湖区扩容,南昌县接壤部分板块或将划入,新建区仅小范围优化?
- 南昌房东必看现在卖房居然不用交个税
- 南昌主流高中男女老师比例(高中)
