期刊名:ACIE
DOI:10.1002/anie.9356220
将二氟甲基(CF₂H)引入药物分子,是调控分子生物活性的常用策略。受强立体电子效应影响,CF₂H 基团可提升分子代谢稳定性、调节亲脂性并增强靶点结合能力。该取代基兼具亲脂性氢键供体与羟基、巯基、氨基等基团的柔性生物电子等排体特性,能够降低上述基团常见的代谢缺陷。因此,开发通用的 CF₂H 引入技术,已成为学术界与工业界的核心目标。现代有机分子直接引入 CF₂H 的方法,多依赖过渡金属催化或介导的有机卤代物与适配 CF₂H 试剂的交叉偶联反应。这类策略已实现芳基卤代物的高效二氟甲基化,但从烷基卤代物出发构建脂肪族 C (sp³)–CF₂H 键的方法仍明显滞后。值得注意的是,富含 sp³ 杂化碳的药物分子通常具有更优的理化与药代动力学性质,临床成功率更高。尽管具备上述优势,向药物分子中直接引入 C (sp³)–CF₂H 基团的进展,仍远落后于芳基类似物。近年来,多种二氟甲基试剂的出现大幅拓展了 C (sp³)–CF₂H 结构的合成空间。羧酸、胺、醇等底物的转化研究取得显著进展,但烷基卤代物的催化二氟甲基化仍局限于烯丙基、苄基、炔丙基、羰基 α- 位等活化底物。沈其龙、刘、Prakash 等课题组的开创性工作表明,非活化烷基卤代物可在特定条件下发生二氟甲基化,但这类方法通常存在底物范围有限(仅兼容烷基碘或一级卤代物)、依赖当量铜盐、需高活性锌试剂或基于硅烷的卤素转移体系等问题。作为卤代物策略的替代方案,二氟甲基自由基向简单烯烃的直接加成,已成为构建脂肪族含 CF₂H 结构的重要途径。这类反应中,CF₂H 自由基通常加成至烯烃末端碳形成稳定烷基自由基,后续可经氢原子转移得到氢二氟甲基化产物,或参与金属催化的双官能化反应。但自由基路径倾向于生成直链产物,高价值支链二氟甲基化异构体的直接选择性合成仍未解决。
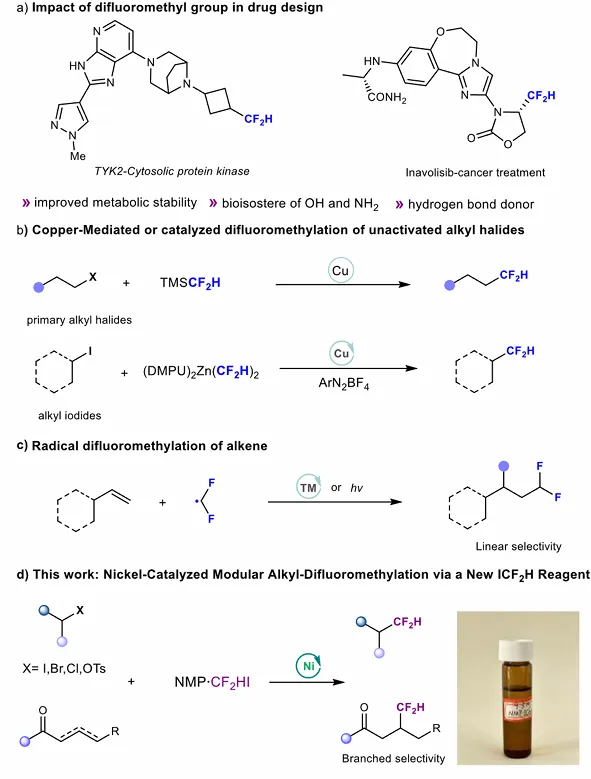
过去数十年,镍催化已实现多种还原交叉偶联反应,包括芳基卤代物与氟烷基亲电试剂偶联形成 C (sp²)–CF₂R 键。相比之下,镍催化烷基卤代物与氟烷基卤代物偶联构建 C (sp³)–CF₂R 键的研究仍未开展。其核心难点在于:烷基卤代物反应性相近,易发生不期望的自身偶联;高价烷基–镍–氟烷基中间体(R–Ni^(III)–CF₂R')受氟烷基强 σ 给电子特性与竞争性 β- 氟消除影响,C (sp³)–C (sp³) 还原消除倾向降低。此外,尽管镍催化烯烃氢官能化反应应用广泛,直接实现氢二氟甲基化以构建 C (sp³)–CF₂H 键的方法仍未实现。鉴于镍催化 C (sp³)–CF₂H 键构建在烷基卤代物与烯烃底物中均存在挑战,研究团队认为,开发一种温和、统一的一步二氟甲基化方法,并采用易获取的 CF₂H 试剂,可有效填补该方法学空白,为药物化学应用提供新可能。
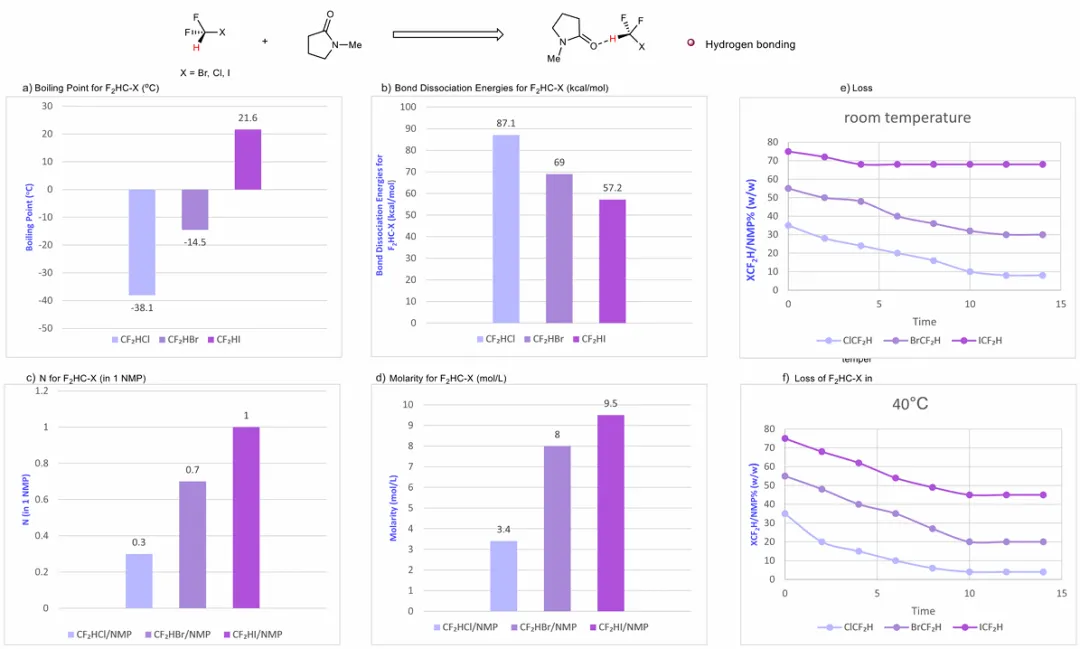
该研究思路将目光投向 CF₂HX(X=Cl、Br、I)试剂 —— 最基础的亲电性二氟甲基源。尽管近年涌现出多种亲电、亲核与自由基型 CF₂H 试剂,多数仍衍生自二氟卤代甲烷(CF₂HX)。CF₂HX 的直接应用受限于常温气态性质(尤其 CF₂HCl 与 CF₂HBr),难以精准控制当量,且在常规有机溶剂中溶解性差。在 CF₂HX(X=Cl、Br、I)中,卤素原子序数增大(Cl<Br<I)伴随沸点升高与碳–卤素(C–X)键解离能降低,表明二氟碘甲烷(CF₂HI)是最适用于镍催化的亲电 CF₂H 试剂。
为解决气态 CF₂HI 的操作与溶解性难题,研究团队通过设计非共价相互作用实现试剂稳定化。CF₂HI 含极化 C–H 键可作为氢键供体,C–I 键可参与次级相互作用。因此,研究团队设想,一种可接受上述相互作用的合适溶剂,能有效稳定并增溶 CF₂HI,最终在室温下得到实用液态试剂。基于该思路,研究团队筛选出 N - 甲基吡咯烷酮(NMP)作为有效介质。NMP 的羰基氧为强氢键受体,可与 CF₂HI 形成稳定 C–H…O 相互作用。密度泛函理论(DFT)计算表明,该氢键复合物为主要作用模式,相互作用能显著、H…O 距离较短,证明氢键强度较高;相比之下,卤素键相互作用显著更弱,在稳定化中作用较小。与该分析一致,溶解实验证实 CF₂HI 与 NMP 形成 1:1 稳定加合物,溶解度高达约 9.5 mol/L,远高于其在四氢呋喃或正己烷中的溶解度。所得 ICF₂H 溶液为便捷液态试剂,可通过注射器精准量取。为评估该稳定化策略的普适性,研究团队考察了 CF₂HBr 与 CF₂HCl。两种试剂与 NMP 的相互作用显著减弱:CF₂HBr 形成约 0.7:1 复合物,CF₂HCl 仅形成 0.3:1 复合物。复合与溶解度趋势为 CF₂HI>CF₂HBr>CF₂HCl,与碘、溴、氯的卤素键供体能力递减规律一致,支持最初的作用假说。ICF₂H 试剂还表现出优异的长期稳定性,数月内制备的多批次样品在 4℃或室温储存后,反应活性无明显差异。研究团队进一步通过常温与高温下的分解监测,评估三种卤代二氟甲基试剂的热稳定性。ICF₂H 在室温下无明显降解,而 CF₂HBr 与 CF₂HCl 均出现明显分解;40℃条件下,ICF₂H 仍为三者中最稳定物种,证实其在储存与操作中的高稳定性。
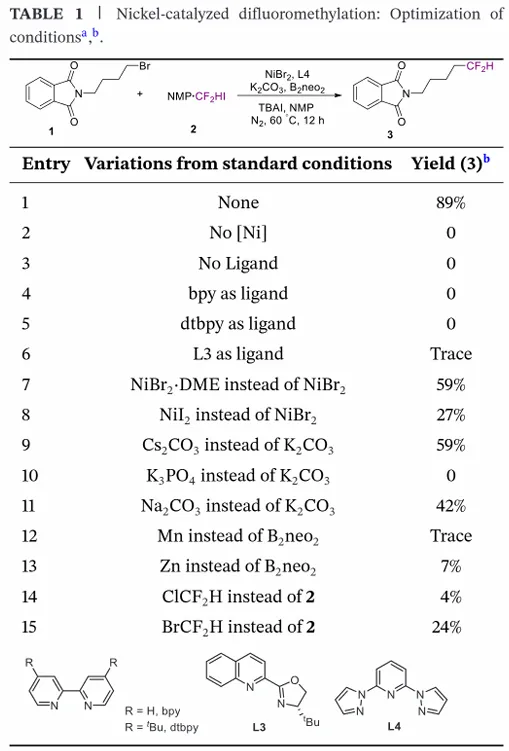
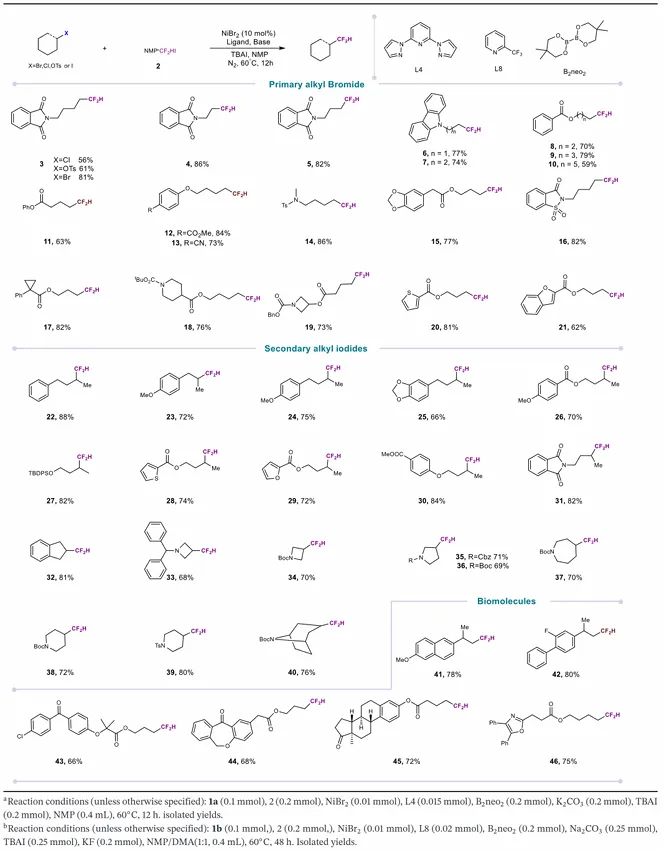
为评估 NMP・CF₂HI 复合物作为实用二氟甲基化试剂的合成潜力,研究团队首先考察其在镍催化非活化烷基溴代物二氟甲基化反应中的表现。以 2-(4 - 溴丁基) 异吲哚啉 - 1,3 - 二酮为模型底物,系统优化催化剂组分与反应参数。在 NiBr₂・DME(10 mol%)、配体 L4(15 mol%)、B₂neo₂(2.0 当量)、K₂CO₃(2.0 当量)、四丁基碘化铵(TBAI,2.0 当量)、NMP(0.4 mL)、60℃条件下,目标产物收率达 89%。对照实验证实,镍催化剂与配体均为必需组分,缺失任一组分均导致反应完全抑制。配体筛选表明,吡啶类双齿配体基本无效,多种氮配体均未得到满意结果。镍源考察显示,NiBr₂效果最优,其他镍 (II) 盐收率显著降低。碱的筛选表明,K₂CO₃为最佳选择,Cs₂CO₃等碳酸盐收率下降,说明 K₂CO₃的碱性与溶解特性最适配该转化。还原剂方面,受龚课题组关于 B₂neo₂在镍催化中高效介导单电子还原的研究启发,研究团队发现该试剂可实现体系有效循环;而常见金属还原剂(锰、锌)仅得到痕量产物或 7% 收率,证实 B₂neo₂在该条件下生成活性镍物种的关键作用。最后,为评估 NMP・CF₂HI 作为二氟甲基源的独特适用性,研究团队在最优条件下对比 CF₂HBr 与 CF₂HCl,两种替代试剂反应性均降低,CF₂HCl 仅得到痕量产物,凸显 CF₂HI 在镍催化中更优的亲电性与活化特性。
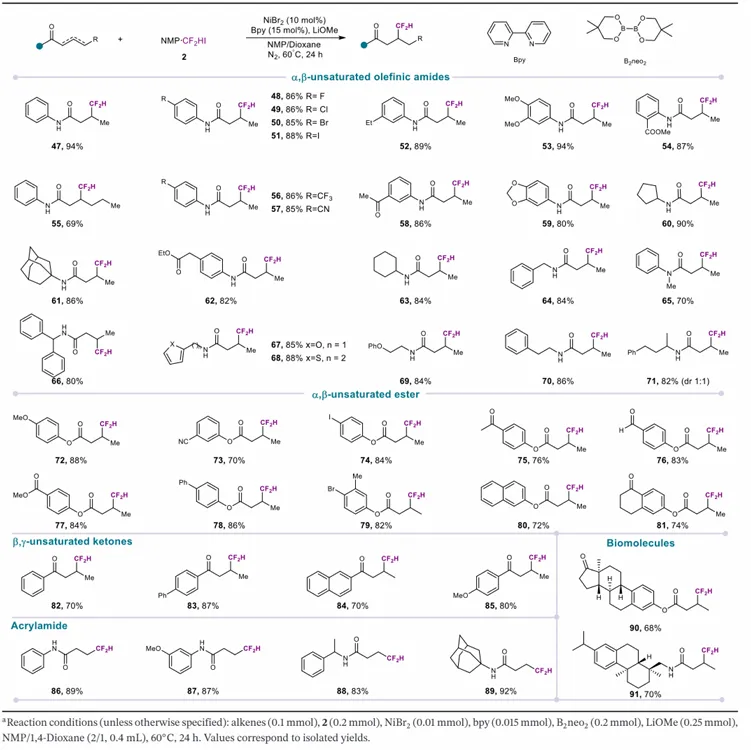
获得最优条件后,研究团队考察该镍催化二氟甲基化反应的底物范围。除烷基溴代物外,烷基氯代物、烷基碘代物、烷基对甲苯磺酸酯均能作为有效亲电试剂,以中等至良好收率得到对应二氟甲基化产物,证明该方法具有广泛的亲电试剂兼容性与实用价值。为系统评估官能团耐受性,研究团队考察多种烷基溴代物。反应在不同电子效应与位阻的底物中均表现出优异稳定性:吸电子基团(邻苯二甲酰亚胺、苯甲酸酯)与给电子胺衍生物均兼容,收率 59%–89%;烷基链长度变化对反应效率影响极小,表明催化体系对碳骨架位阻变化不敏感。该方法还兼容多种酯衍生物(苯氧酯、胡椒基、苯丙酸、哌啶),收率良好至优秀;醚类与保护胺也能兼容, consistently 得到高收率产物。上述结果共同证明,反应路径可耐受取代基的电子与位阻变化。鉴于杂环在药物化学中的普遍性,研究团队进一步考察多种杂环底物:苯并异噻唑、氮杂环丁烷、噻吩、苯并呋喃均能兼容反应条件,收率 62%–82%。最后,该方法的合成实用性通过药理活性化合物的后期二氟甲基化得到验证:萘普生、氟比洛芬、非诺贝酸、伊索克酸、雌酮、奥沙普秦均能良好兼容,收率 66%–80%。上述结果确立该方法为复杂类药分子后期引入二氟甲基的实用、通用策略。
受一级烷基溴代物成功反应的鼓舞,研究团队尝试将该转化拓展至二级烷基溴代物,但发现反应收率较低,遂以二级烷基碘代物为底物系统优化条件。后续研究表明,2 - 三氟甲基吡啶为配体时,可在镍催化条件下实现与 NMP・CF₂HI 的高效偶联,得到目标烷基二氟甲基化产物;而联吡啶、邻菲啰啉等其他双齿配体效率较低。理性优化确定 B₂neo₂/Na₂CO₃为最优还原剂–碱组合。条件确立后,研究团队考察二级烷基碘代物的底物范围:多种结构多样的衍生物均能兼容反应,含芳基、富电子芳基、酯基、醚基的底物均顺利发生二氟甲基化,收率良好至优秀;含酸敏感硅基保护基的底物也能有效反应,证明条件温和可保护敏感官能团。杂环底物与邻苯二甲酰亚胺衍生物也能成功转化。该方法还兼容多种含氮杂环(环丁烷、哌啶、氮杂环丁烷、氮杂环庚烷骨架),均以良好至优秀收率得到对应二氟甲基化产物。值得注意的是,尽管二级烷基碘代物底物范围广泛,叔烷基卤代物在标准条件下不兼容。
烯烃作为基础大宗化工原料与多功能官能团转化平台,其氢二氟甲基化反应的开发仍具挑战性。基于上述烷基亲电试剂镍催化二氟甲基化的成功,研究团队尝试将该催化体系拓展至非活化烯烃的氢二氟甲基化。经系统考察与大量反应参数优化,最终得到该转化可靠、通用的反应方案。在最优条件下,研究团队考察该镍催化非活化烯烃氢二氟甲基化的底物范围:反应在多种 α,β- 不饱和烯烃酰胺中应用广泛,芳环取代基电子性质对反应结果影响较小;多种含给电子或吸电子基团的芳基底物均顺利发生二氟甲基化,收率中等至良好。氟、氯、溴、碘等卤素取代基也能良好兼容,不降低反应效率,凸显该转化的稳定性与合成实用性。多种脂肪族酰胺也适用于该反应:环状底物与无环烷基链均高效发生二氟甲基化;底物范围还涵盖杂芳环体系、N - 保护衍生物、末端烯烃含长碳链的酰胺。α,β- 不饱和酯也能兼容,收率 77%–88%;β,γ- 不饱和酮与丙烯酰胺也能有效参与反应,得到合成实用的收率。最后,该方法的实用性通过复杂生物活性分子的后期修饰得到验证:雌酮与 PDK 抑制剂烈辣明衍生的高密度官能化衍生物均能在标准条件下成功转化。上述结果共同证明,该氢二氟甲基化方案具有优异的官能团耐受性与广泛适用性。
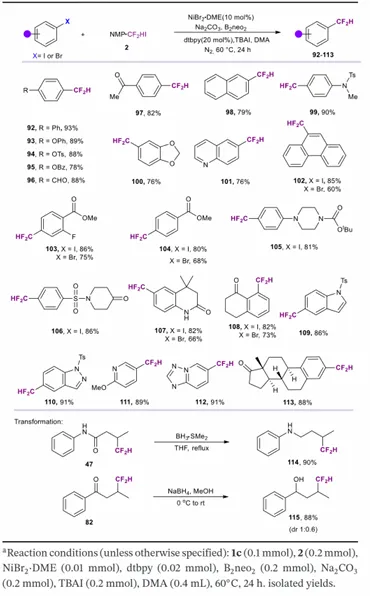
基于上述烷基亲电试剂二氟甲基化与非活化烯烃氢二氟甲基化的镍–硼催化体系成功开发,研究团队尝试将该统一平台拓展至芳基卤代物。令人欣喜的是,该催化体系对芳基碘代物的二氟甲基化效果广泛,可在温和条件下直接得到二氟甲基化芳烃。通常,含给电子与吸电子取代基的芳基碘代物均对 ICF₂H 表现出良好反应性,收率中等至良好;苯基、萘基、菲、烷基苯氧基均兼容,得到目标 CF₂H 产物。.
反应还兼容醛、酮、酯等亲电性官能团。重要的是,含哌嗪、哌啶酮、喹啉酮、喹啉、吲哚、吡啶等杂环基团的底物,也能以中等至良好收率得到目标产物。此外,该方法可实现雌酮的后期二氟甲基化。研究团队进一步考察芳基溴代物在该条件下的反应性:苯环含强吸电子基团时,反应顺利进行得到目标产物,收率略低于对应芳基碘代物;而芳环含给电子基团时仅观察到痕量产物。遗憾的是,芳基氯代物在该转化中完全不反应。
为展示二氟甲基化产物的合成多功能性,研究团队开展代表性下游衍生化研究:硼烷–二甲基硫醚还原化合物 47 的羰基得到化合物 114,硼氢化钠还原化合物 82 得到对应醇 115。上述转化证明,二氟甲基化中间体可作为灵活合成砌块用于进一步衍生。
为深入理解二氟甲基化反应机理,研究团队开展一系列对照实验。首先,以四甲基哌啶氮氧化物(TEMPO)进行自由基捕获实验:向含底物 1 与 ICF₂H 的标准反应体系中加入 TEMPO(4.0 当量),产物生成完全抑制,气相色谱–质谱(GC-MS)检测到对应 TEMPO 加合物。为进一步支持自由基中间体参与,开展自由基时钟实验:自由基时钟底物 3b 在反应条件下得到 15% 收率的开环产物 S2,原料未完全消耗。随后,考察原位生成烷基硼 neo 中间体 / 铃木型偶联路径的可能性:标准条件下,烷基硼 neo 与 ICF₂H 2 的交叉偶联未得到目标产物,表明烷基硼 neo 不参与还原交叉偶联过程,铃木型偶联序列可能性较低。研究团队还考察镍催化剂的作用:标准条件下以镍 (0) 替代镍 (II) 前体,未得到目标产物 3,表明镍 (0) 并非催化相关活性物种。为探究 Ni–CF₂H 中间体的可行性,开展当量实验:镍 (0) 与镍 (II) 混合物原位生成镍 (I),低温下与 (DMPU)₂Zn (CF₂H)₂反应原位生成 Ni^(I)–CF₂H 物种,随后加入烷基溴代物,60℃搅拌 24 h,以 42% 收率得到目标交叉偶联产物。该结果证明,Ni^(I)–CF₂H 物种为化学可行中间体,可与烷基溴代物发生有效偶联得到对应二氟甲基化产物,强力支持所提出的反应机理。
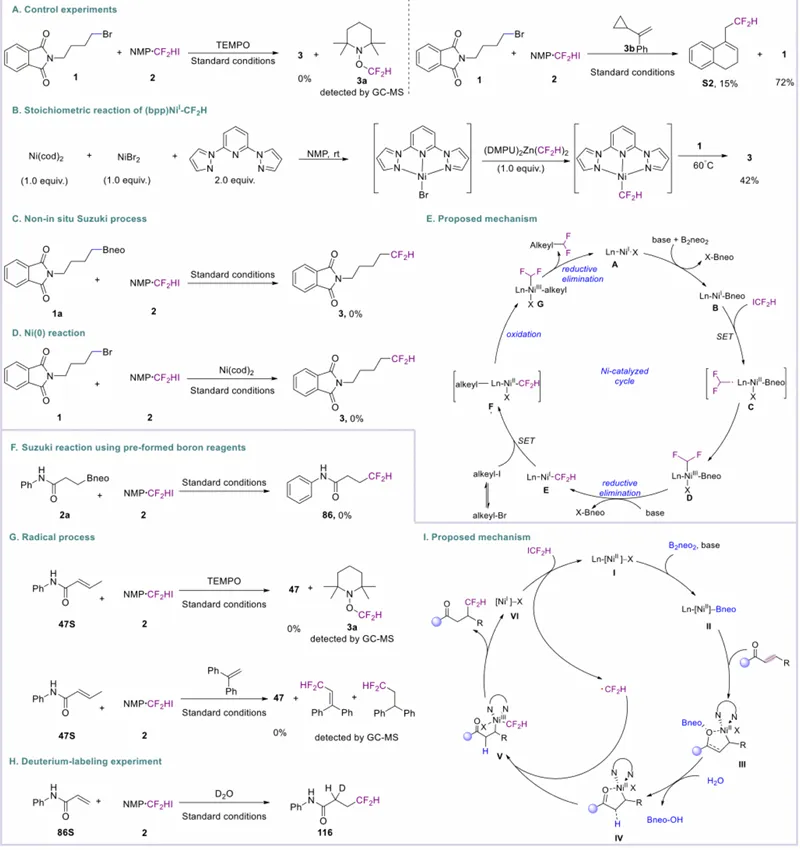
基于上述机理研究与文献先例,研究团队提出烷基卤代物二氟甲基化的催化循环:催化循环始于镍 (I) 配合物 A 的硼基化,生成 Bneo–镍 (I) 物种 B;物种 B 与 CF₂H–I 发生单电子转移(SET),得到二氟甲基自由基与镍 (II) 中间体 C;随后自由基氧化加成得到镍 (III) 物种 D,经 Bneo–X 消除得到镍 (I) 中间体 E;此时烷基溴代物可与中间体 E 顺利反应,生成烷基自由基与 Rf–镍 (II) 物种 F;烷基自由基与 F 快速重组得到镍 (III) 中间体 G,最终经还原消除得到目标偶联产物并再生镍 (I) 催化剂 A。同时,研究团队也探究了烯烃二氟甲基化的反应机理:首先,烷基硼试剂 2a 在标准条件下未得到目标产物,表明烷基硼 neo 不参与还原交叉偶联过程。类似地,向含 47 与 ICF₂H 的体系中加入 TEMPO(2.0 当量),仅生成 TEMPO 加合物,未观察到目标产物,表明反应经由自由基路径进行。自由基时钟实验进一步支持该推测。最后,以 D₂O 开展氘代标记实验,证实产物中一个氢原子来源于溶剂中的微量水。
基于上述机理观察,研究团队提出非活化烯烃氢二氟甲基化的合理催化循环:催化循环始于镍 (II) 前体与大位阻硼烷试剂 B₂neo 在碱存在下发生配体交换,形成镍 (II) 配合物 I;同时,非活化烯烃被碱去质子化并发生共轭 1,4 - 加成,生成羰基导向的五元镍环中间体 III;随后,中间体 III 在碱与水存在下发生脱硼羟基化,释放 Bneo–OH 并促进配体氧化加成,得到对应镍 (II) 物种。该脱硼羟基化步骤被认为经由烷基硼酸酯 / 水复合物形成进行。单电子转移生成 CF₂H 自由基,被镍 (III) 中心捕获形成镍 (III)–CF₂H 配合物 V;V 经还原消除得到氢二氟甲基化产物与镍 (I) 物种 VI;最后,物种 VI 被再氧化再生镍 (I) 前体 I,完成催化循环。
综上,本文开发一种温和、通用的镍–硼协同催化平台,可实现烷基卤代物、芳基与烯基卤代物以及非活化烯烃的二氟甲基化反应。该策略采用 N - 甲基吡咯烷酮(NMP)稳定化的二氟碘甲烷(ICF₂H)为试剂,NMP 作为氢键受体介质,克服了试剂操作与溶解性的长期难题,使 ICF₂H 可在高有效浓度下应用。该催化体系对多种官能团、杂环与复杂分子结构具有广泛底物耐受性,可便捷应用于生物活性化合物的后期二氟甲基化。机理研究支持涉及镍 (I/II/III) 中间体的自由基路径,与镍–硼介导的活化模式一致。总体而言,该工作建立了一种统一、实用的二氟甲基引入方法,适用于多类亲电试剂与不饱和骨架,为药物相关骨架的合成提供了通用平台。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
