南昌大学陈义旺教授&谈利承教授团队Nature.Ener.:通过有机铯盐在α-FA-Cs钙钛矿太阳能电池中可控掺入Cs+并获26.61%认证效率
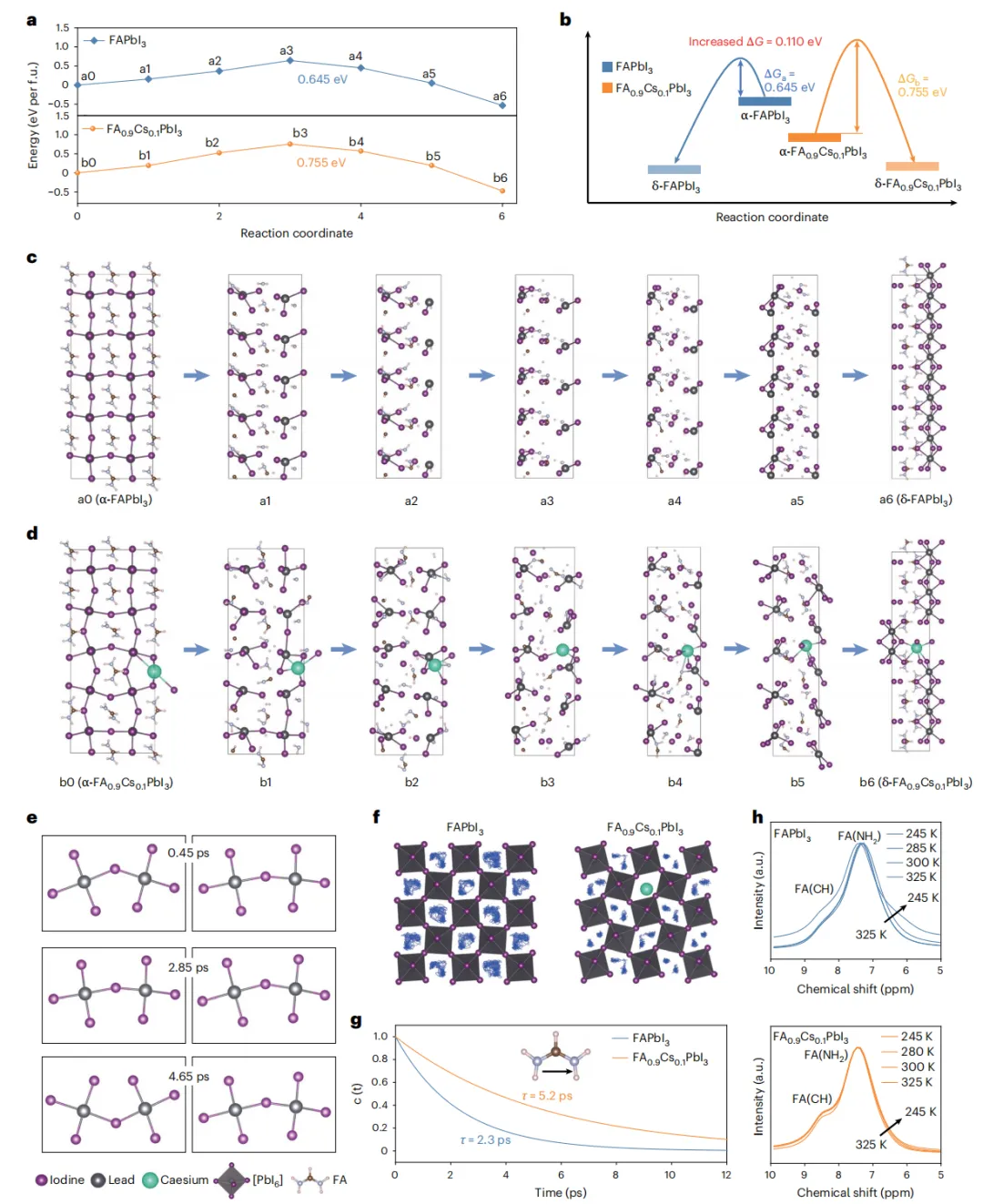
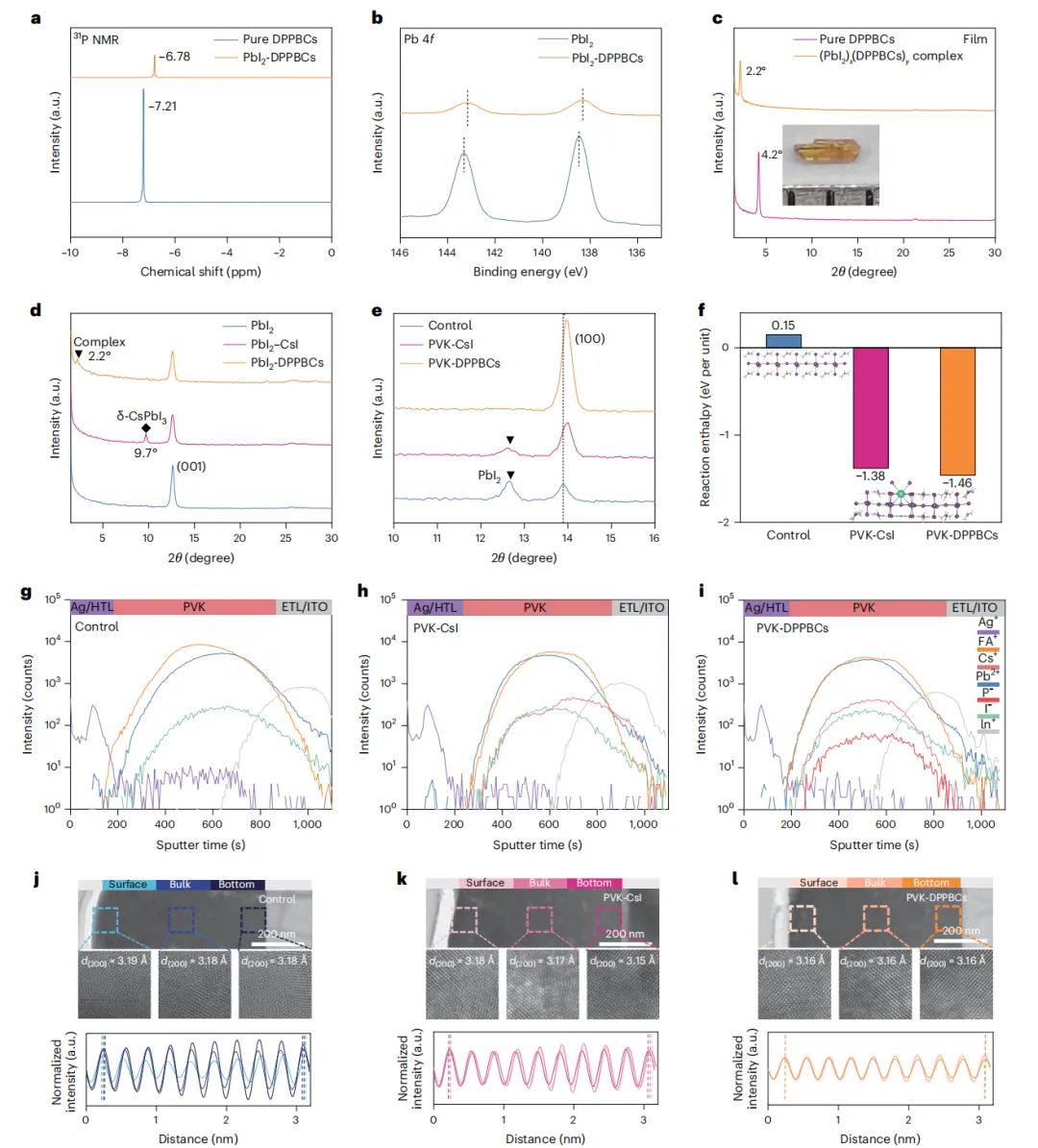
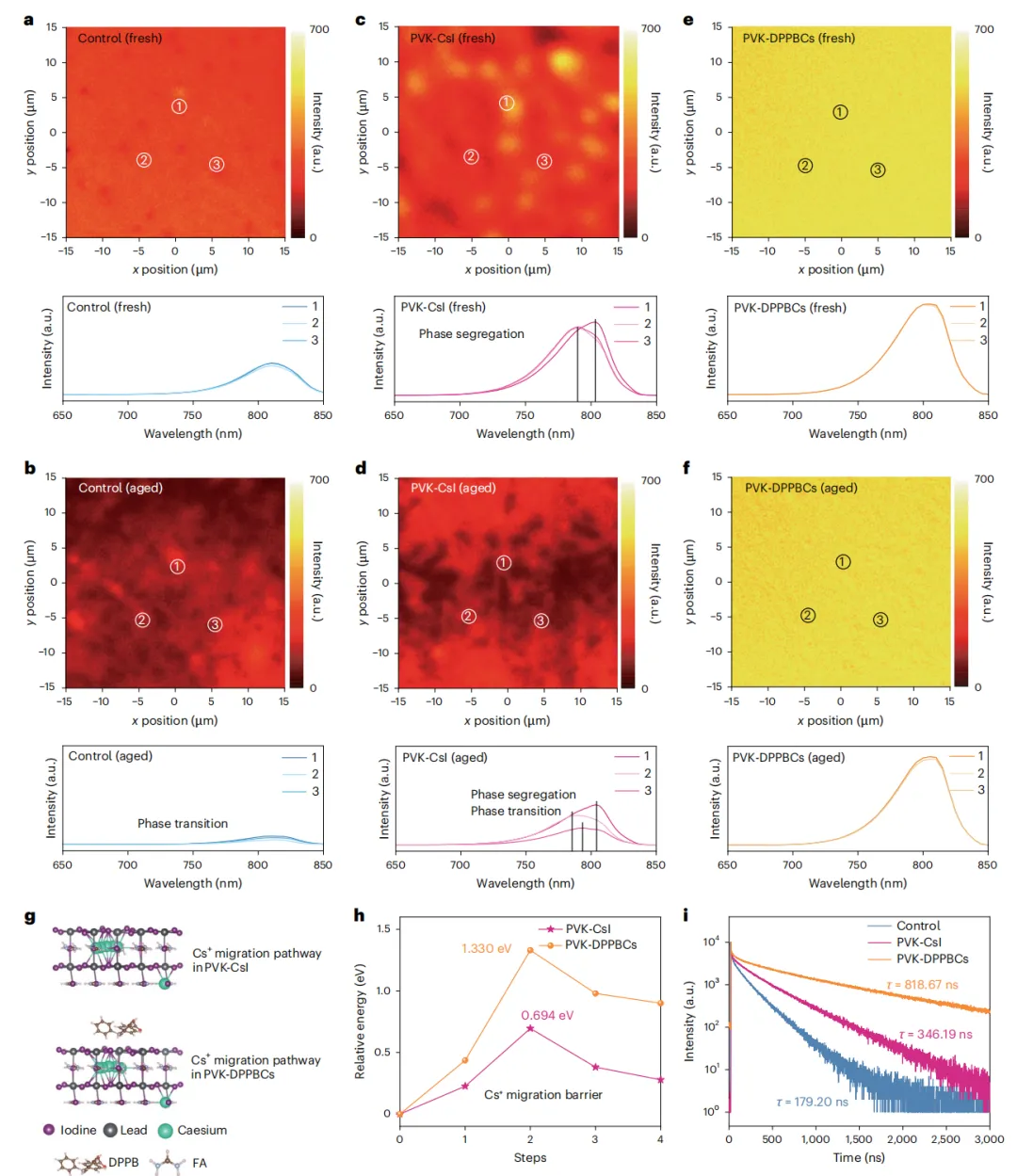
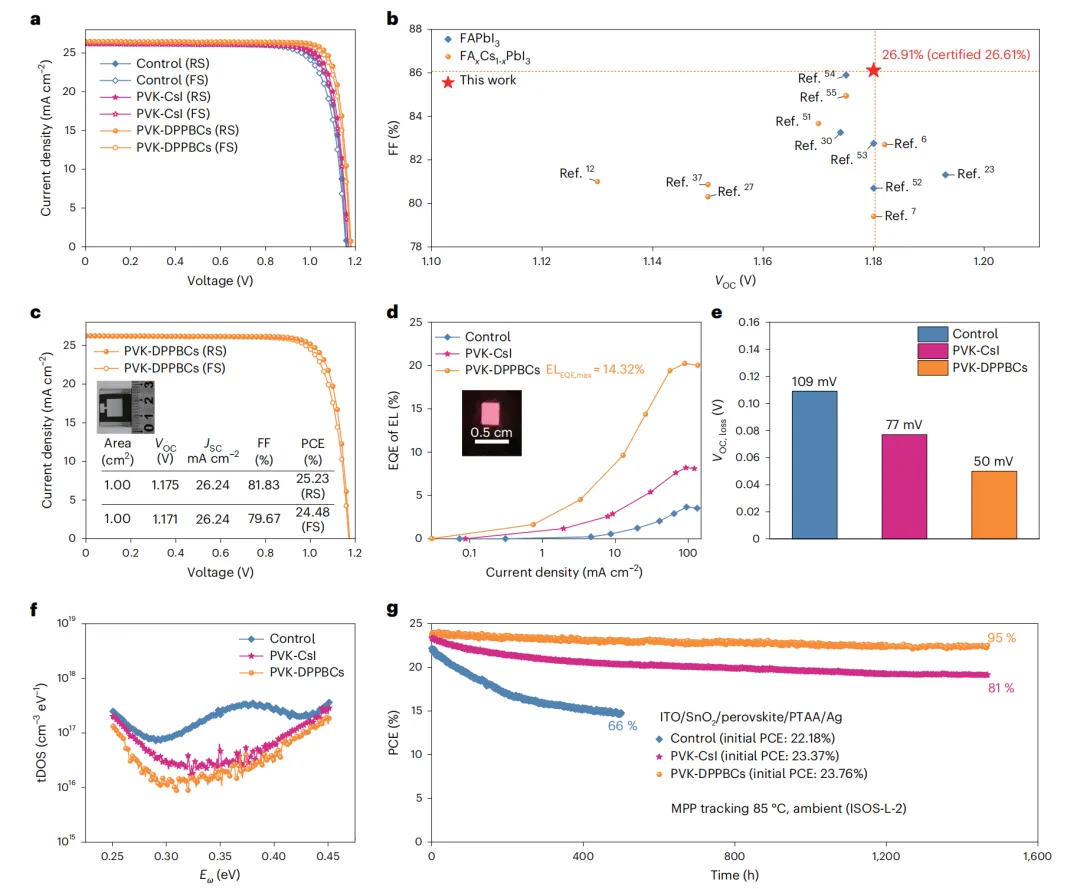
本文直接翻译论文摘要如下:两步法制备的甲脒-铯金属卤化物钙钛矿(FA1-xCsxPbI3)能够为结晶调控提供卓越的控制,因此更适用于太阳能电池的制造。然而,由于有限的Cs+掺入和不清的相变机制,要形成稳定器件所需的理想α相仍然具有挑战性。在此,我们设计了4-(二苯基膦)苯甲酸铯,以实现高效的Cs+掺杂并使阳离子分布均匀化,从而获得具有更高相稳定性的高质量钙钛矿薄膜。结果,通过两步法制备的太阳能电池实现了26.91%(认证26.61%)的效率。集成了热稳定电荷传输层的器件在85°C、1个太阳光照射、最大功率点跟踪的连续运行1500小时(ISOS-L-2协议)后,仍能保持其初始效率(23.76%)的95%。本研究深入揭示了FA0.9Cs0.1PbI3的相变路径及相应的过渡态结构,以及Cs+驱动的钙钛矿晶格稳定化机制。深入剖析 :这项工作针对两步法钙钛矿太阳能电池产业化中的核心难题——Cs+均匀、高效掺入以稳定α相,提出了一种创新的分子工程策略。研究者合成的有机铯盐DPPBCs,其有机部分(膦基苯甲酸根)能够与PbI2前驱体强效配位,不仅抑制了有害δ-CsPbI3相的生成,还促进了Cs+在钙钛矿晶格中的均匀分布。这从源头上改善了薄膜质量,提升了相稳定性。更重要的是,该工作通过理论计算与实验结合,首次从动力学和热力学角度,在原子层面阐明了Cs+如何通过“锁定”[PbI6]4-八面体的振动和限制FA+阳离子的旋转,来提升FA-Cs钙钛矿的晶格稳定性。最终实现的26.61%认证效率是两步法FA-Cs基钙钛矿太阳能电池的最高水平之一,而其展示的在高温、光照下的卓越运行稳定性,极大地推进了此类电池的商业化应用前景。有机-无机杂化钙钛矿太阳能电池因其卓越的光伏性能和简单的溶液加工优势,被视为可持续能源领域的一颗新星,其功率转换效率已超过26.7%。然而,器件的稳定性问题仍是其商业化应用的关键挑战。在众多制备工艺中,两步法(先沉积PbI2,再与有机铵盐反应)由于能够将成核与晶体生长过程解耦,提供了更好的晶体调控能力、均匀性和可重复性,尤其适合规模化生产,展现出巨大的商业化潜力。目前,基于两步法制备的高性能钙钛矿太阳能电池主要是甲脒(FA)基体系。然而,纯α-FAPbI3在室温下会自发转变为非光活性的δ相,严重损害了器件的稳定性。通过A位阳离子合金化形成FA1-xCsxPbI3钙钛矿被认为是增强器件稳定性的有效策略。但在两步法工艺中实现高性能的FA-Cs基电池仍面临严峻的技术障碍,这主要归因于CsX在异丙醇中的低溶解度,以及将其直接掺杂到PbI2前驱体溶液中时会生成δ-CsPbI3相,导致钙钛矿薄膜组分不均匀,并限制了Cs+的有效掺入。因此,探索在两步法中有效引入Cs+同时避免相分离的方法,对于提升器件的运行稳定性至关重要。此外,目前关于钙钛矿相变的研究主要基于纯FAPbI3结构模型,Cs+如何提高钙钛矿稳定性的机制尚不清楚。钙钛矿具有物理上的软晶格和高动态特性,[PbI6]4-八面体的Pb-I-Pb键角在其平衡位置附近振荡,A位阳离子在[PbI6]4-八面体笼内持续旋转。因此,深入理解Cs+掺杂对FA1-xCsxPbI3钙钛矿相变热力学和动力学的影响至关重要。为解决上述问题,在本工作中,我们合成了一种有机铯盐——4-(二苯基膦)苯甲酸铯(DPPBCs)。该盐能够在两步法制备的钙钛矿中实现A位Cs+的有效掺杂,并促进钙钛矿薄膜中A位阳离子的均匀分布。这种均匀化作用抑制了相分离和不期望的相变。此外,我们利用密度泛函理论计算,阐明了FA0.9Cs0.1PbI3的相变路径及相应的结构变化,同时研究了Cs+对相变动力学和热力学驱动力的影响。我们的分析表明,Cs+通过“锁定”[PbI6]4-骨架,减少了Pb-I-Pb振荡并限制了FA+的旋转。同时,DPPBCs增加了FA0.9Cs0.1PbI3相变的能垒。最终,经DPPBCs处理的钙钛矿太阳能电池实现了26.91%(认证26.61%)的效率。采用热稳定电荷传输层的太阳能电池在85°C、1个太阳光照射、最大功率点跟踪条件下连续运行1500小时(ISOS-L-2协议)后,仍能保持其初始效率(23.76%)的95%。本研究为理解FA0.9Cs0.1PbI3中α-δ相变的热力学和动力学提供了见解,并为钙钛矿太阳能电池的设计提供了指导。为了研究Cs+对增强FA-Cs钙钛矿晶格稳定性的影响,研究者通过DFT计算对比了FAPbI3和FA0.9Cs0.1PbI3的相变过程。对于FAPbI3,相变始于[PbI6]4-八面体的旋转,导致Pb-I键断裂和I离子迁移(能垒0.645 eV),进而从立方α相的三维角共享结构转变为六方δ相的一维面共享链状结构。而在FA0.9Cs0.1PbI3中,相变过程中观察到的Pb-I键断裂更少,其热力学相变能垒更高(0.755 eV)。分子动力学模拟显示,Cs+的掺入“锁定”了[PbI6]4-八面体的振动,显著降低了Pb-I-Pb键角的振荡幅度,并限制了FA+阳离子的旋转自由度(从几乎无限制的三维旋转变为特定方向的局域运动)。固体核磁共振结果也证实,FA0.9Cs0.1PbI3中FA+的重定向动力学受到限制。这些结果表明,Cs+通过增强晶格刚性、限制有机阳离子运动,从而提高了相变能垒和热力学稳定性。为了在两步法中有效引入Cs+并避免δ-CsPbI3杂质相,研究者合成了有机铯盐DPPBCs。其分子中的P原子作为路易斯碱,能与PbI2中未配位的Pb原子强效结合,形成(PbI2)x(DPPBCs)y配合物薄膜。XRD证实,在传统的PbI2-CsI薄膜中出现了δ-CsPbI3相(9.7°),而在PbI2-DPPBCs薄膜中该杂相消失,并出现了对应于配合物的新衍射峰(2.2°)。这证明DPPBCs通过配位作用抑制了δ-CsPbI3的生成。在随后的钙钛矿转化过程中,DFT计算表明,引入DPPBCs的体系具有最低的反应焓(-1.46 eV per unit),有利于降低反应能垒,促进晶体生长。最终获得的PVK-DPPBCs钙钛矿薄膜不仅实现了Cs+的有效掺入(XRD角位移证明),而且FA+和Cs+的分布非常均匀(飞行时间二次离子质谱ToF-SIMS深度剖面证实),而对照组和PVK-CsI组则分别存在FA分布不均和Cs+在底部富集的问题。高角环形暗场扫描透射电镜(HAADF-STEM)也显示PVK-DPPBCs薄膜的晶面间距(d(200))变化可忽略,表明其均匀的阳离子分布释放了晶格应力。离子在薄膜中的异质分布会导致严重的相分离,影响器件寿命。二维荧光成像显示,在环境空气中连续光照48小时后,对照薄膜的荧光强度因相变而急剧下降,PVK-CsI薄膜的荧光峰发生分裂,表明存在相分离。而PVK-DPPBCs薄膜的荧光强度和峰位均无明显变化,表现出优异的相稳定性。DFT计算进一步揭示,由于DPPBCs与钙钛矿的配位作用,PVK-DPPBCs薄膜中Cs+和I-的迁移能垒显著高于对照组和PVK-CsI组,这有利于抑制离子迁移引发的相分离和相变。瞬态吸收和开尔文探针力显微镜测试也证实,PVK-DPPBCs薄膜具有更长的载流子寿命和更低的表面电子陷阱密度。基于n-i-p平面结构(ITO/SnO2/钙钛矿/Spiro-OMeTAD/Ag)的冠军器件性能如下表所示:器件 | 扫描方向 | Voc (V) | Jsc (mA cm⁻²) | FF (%) | PCE (%) | 迟滞因子 (%) |
|---|
对照 | 反向 | 1.161 | 26.35 | 81.26 | 24.86 | 2.8 |
| 正向 | 1.158 | 26.27 | 79.42 | 24.16 | |
PVK-CsI | 反向 | 1.168 | 26.22 | 83.53 | 25.56 | 2.3 |
| 正向 | 1.167 | 26.22 | 81.63 | 24.98 | |
PVK-DPPBCs | 反向 | 1.180 | 26.48 | 86.11 | 26.91 | 2.0 |
| 正向 | 1.180 | 26.45 | 84.48 | 26.36 | |
PVK-DPPBCs器件获得了26.91%的最高效率(认证效率26.61%)和86.11%的高填充因子,其稳态输出效率约为26.51%。效率的提升主要归因于薄膜质量的改善、缺陷的减少以及电荷载流子提取能力的增强。该器件在作为发光二极管(LED)测试时,也显示出最高的电致发光效率(14.32%)和最低的电压损失(50 mV),表明其缺陷相关的非辐射复合被极大抑制。陷阱态密度(tDOS)谱显示,PVK-DPPBCs器件在几乎整个能级深度区域都具有更低的陷阱密度。稳定性测试表明,PVK-DPPBCs钙钛矿薄膜在光照2000小时后无明显降解,且具有优异的热稳定性。更重要的是,当使用热稳定性更好的空穴传输材料PTAA替代Spiro-OMeTAD并封装后,PVK-DPPBCs器件在85°C、1个太阳光照射、最大功率点跟踪的苛刻条件(ISOS-L-2协议)下连续运行1500小时后,仍能保持其初始效率(23.76%)的95%。这一显著的稳定性提升归因于DPPBCs有效抑制了钙钛矿的相分离/相变,同时实现了Cs+的高效均匀掺杂。本研究阐明了FA0.9Cs0.1PbI3钙钛矿的相变路径及相应的结构变化,结论指出Cs+通过“锁定”[PbI6]4-八面体振动和抑制FA+旋转,增强了晶格稳定性并提高了相变能垒。同时,通过在FA0.9Cs0.1PbI3的两步法制备过程中引入有机铯盐DPPBCs,成功抑制了δ-CsPbI3的生成,并实现了Cs+的有效掺杂。DPPBCs的引入降低了钙钛矿形成的反应焓,促进了Cs+和FA在钙钛矿薄膜中的均匀分布,从而增强了相稳定性。最终,经DPPBCs处理的太阳能电池获得了26.91%的冠军效率(认证26.61%),其Voc为1.18 V,FF高达86.11%。此外,采用热稳定电荷传输层的封装器件在85°C、1个太阳光照射下连续运行1500小时后,仍能保持95%的初始效率。这些发现为理解FA-Cs钙钛矿的相分离/相变提供了深刻见解,并为优化长期稳定的FA-Cs钙钛矿太阳能电池提供了科学指导。图1、FAPbI3和FA0.9Cs0.1PbI3相变过程的动力学和热力学分析。a, b,FAPbI3 (a) 和FA0.9Cs0.1PbI3 (b) 从α相到δ相相变过程的DFT计算能垒。c, d,FAPbI3 (c) 和FA0.9Cs0.1PbI3 (d) 从α相到δ相相变过程中对应的晶体结构演变。e,FAPbI3和FA0.9Cs0.1PbI3中Pb-I-Pb键角随时间变化的轨迹。f,FA+中氮原子位置随时间累积的密度分布图。g,FA+阳离子的矢量自相关函数,表示FA+保持其初始取向随时间的概率。插图:用箭头表示的FA+离子分子矢量。h,FAPbI3和FA0.9Cs0.1PbI3晶体的变温固态¹H NMR谱。图2、DPPBCs实现Cs+高效掺杂的机制。a, DPPBCs的分子结构。b, CsI (左) 和DPPBCs (右) 在两步法过程中引入Cs+的示意图。c, (PbI2)x(DPPBCs)y配合物的晶体结构。d, PbI2、PbI2-CsI和PbI2-DPPBCs薄膜的XRD图谱。e, 对照、PVK-CsI和PVK-DPPBCs钙钛矿薄膜的XRD图谱。f, 对照、PVK-CsI和PVK-DPPBCs样品形成钙钛矿的反应焓计算。g-i, 对照(g)、PVK-CsI (h) 和PVK-DPPBCs (i) 样品的ToF-SIMS深度剖面。HTL,空穴传输层,Spiro-OMeTAD;ETL,电子传输层,SnO2。j-l, 上:对照(j)、PVK-CsI (k) 和PVK-DPPBCs (l) 样品的截面HAADF-STEM图像。下:图展示了计算强度的变化,计算的面间距在对应图像中给出。d(200) 代表钙钛矿的(200)晶面间距。图3、钙钛矿薄膜在光照条件下的稳定性。a-f, 在环境空气中连续光照老化48小时前后,对照(a, b)、PVK-CsI (c, d) 和PVK-DPPBCs (e, f) 钙钛矿薄膜的二维荧光强度分布图(上)及从选定区域提取的荧光光谱(下)。g, h, PVK-CsI和PVK-DPPBCs钙钛矿晶格中I⁻离子迁移路径(g)和Cs⁺离子迁移能垒(h)。i, 对照、PVK-CsI和PVK-DPPBCs样品的瞬态荧光光谱。图4、器件性能与稳定性。a, 基于Spiro-OMeTAD的冠军器件的电流密度-电压曲线。b, 本研究与其他报道的高效两步法FA-Cs基钙钛矿太阳能电池的填充因子和效率统计图。c, 有效面积为1.00 cm²的PVK-DPPBCs器件的电流密度-电压曲线。d, 对照、PVK-CsI和PVK-DPPBCs器件在LED工作模式下的电致发光量子效率。插图:PVK-DPPBCs器件的电致发光图像。e, f, 对照、PVK-CsI和PVK-DPPBCs器件的电压损失分析(e)和陷阱态密度谱(f)。E是分界能量。g, 采用PTAA作为空穴传输层的封装对照、PVK-CsI和PVK-DPPBCs器件,在连续1个太阳光等效白光LED照射、最大功率点跟踪、遵循ISOS-L-2协议(85°C,环境湿度)条件下的效率演化。作者 :Jiacheng He1,2,5, Zhao Guo1,5, Kaikai Liu1, Wangping Sheng1, Xiao Luo1, Licheng Tan1 & Yiwang Chen1,3,4- Licheng Tan *:College of Chemistry and Chemical Engineering/Film Energy Chemistry for Jiangxi Provincial Key Laboratory/Institute of Polymers and Energy Chemistry, Nanchang University, Nanchang, China. (e-mail: tanlicheng@ncu.edu.cn)
- Yiwang Chen *:1 College of Chemistry and Chemical Engineering/Film Energy Chemistry for Jiangxi Provincial Key Laboratory/Institute of Polymers and Energy Chemistry, Nanchang University, Nanchang, China. 3 Key Laboratory of Fluorine and Silicon for Energy Materials and Chemistry of Ministry of Education, Jiangxi Normal University, Nanchang, China. 4 College of Chemistry and Chemical Engineering, Gannan Normal University, Ganzhou, China. (e-mail: ywchen@ncu.edu.cn)
https://doi.org/10.1038/s41560-026-02016-7本文来源各大出版社论文数据库,版权归文章出版社所有;本文内容采用AI辅助整理生成,如有错漏请私信联系;本文仅用于学术分享,转载请注明出处;如需推广本人学术成果和商务合作请私信联系,若有错漏或侵权请私信联系删除或修改!
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
